简介
ChIP-Seq:用于在全基因组范围中研究DNA结合蛋白(相互反应)、组蛋白修饰(表观遗传标记)和核小体的技术,研究这三个主题可有助于了解基因之间的相互调控以及染色体的功能结构。
ChIP-Seq实验原理:示意图为Fig.1和Fig.2. 在生理状态下,把细胞内的DNA与蛋白质交联(Crosslink)后裂解细胞,分离染色体,通过超声或酶处理将染色质随机切割,利用抗原抗体的特异性识别反应,将与目的蛋白相结合的DNA片段沉淀下来,再通过反交联(Reverse crosslink)释放结合蛋白的DNA片段,最后测序获得DNA片段的序列。
注意:当研究重点是得到核小体的位置和组蛋白修饰的位置的时候,实验中并不首先进行crosslink,而是用超声或者MNase直接进行打断,优先使用MNase,可以更高效地去除掉linker DNA片段以得到核小体更为精确的位置。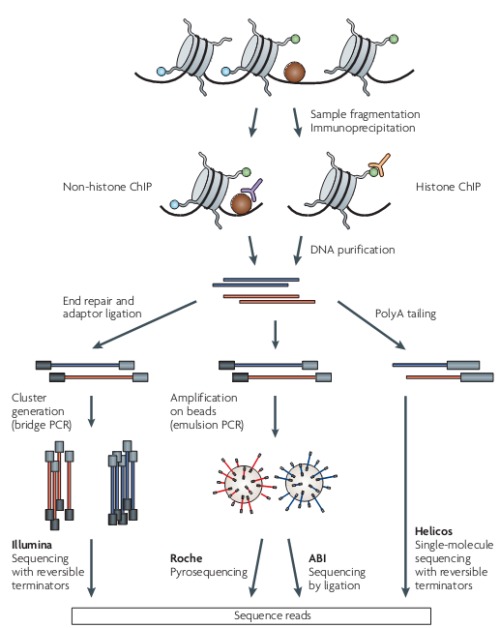
Fig.1
Fig.2
ChIP-Seq的优势:
1. 具有碱基层面的分辨率;
2.不会有ChIP-chip中由DNA片段杂交导致的噪音,GC含量、片段长度、片段浓度以及耳机结构都会对杂交造成影响;
3.ChIP-chip中的微阵列信号不是线性增长的,其所测量的范围有限。
4. 由于在设计array时,探针的数量、种类有限,当coverage比较高的时候无法准确测量,也无法发现新的序列。
ChIP-Seq与ChIP-chip的比较见Fig.3.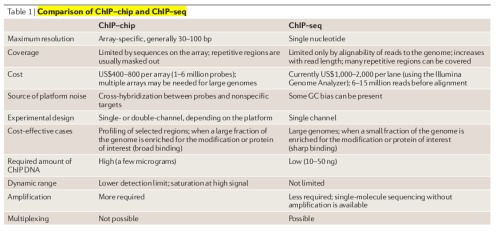
Fig.3
实验设计的关键
抗体质量:一个灵敏度高和特异性高的抗体可以得到富集的DNA片段,这有利于探测结合位点。
样本量:Illumina,10-50ng DNA,需要用PCR进行扩增的轮数也比较少,因而由PCR导致的偏差比较小
空白对照:空白对照是必要的,存在很多假阳性情况,举例:1. 开放的染色体区域更容易被打断成片段,这样导致tag数在基因组上的分布是不均匀的;2.很多重复序列会使做map的时候得到结果难以解释。空白对照的用途:可以判断由ChIP-Seq得到的peak时候具有统计上的显著性。
三种类型的空白对照:1.部分进行免疫共沉淀前的DNA(input DNA),这是最常用的;2.由免疫共沉淀得到而不含有抗体的DNA(mock IP DNA),使用这个的一个问题是收集到的量可能不够;3.使用非特异免疫共沉淀方法得到的DNA.
测序深度:在发表的ChIP-Seq实验中,一般使用Illumina Genome Analyzer上一个lane产生的数据作为一个基本单位,目前一个lane大概是8-15million reads(2009数据)。判断足够的测序深度的标准是:当增加测序,得到更多的reads的时候不能发现更多的东西。应该这一准则到结合位点的数量上就是:进行测序,增加reads数而无法得到更多的结合位点。
关于测序深度饱和曲线的讨论见:Kharchenko, P.V., Tolstorukov, M.Y. & Park, P.J. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nature biotechnology 26, 1351-1359 (2008).
饱和曲线示例: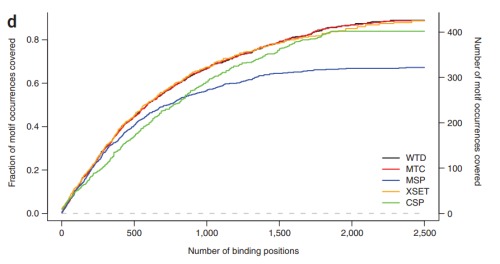
Multiplexing:对于基因组比较小的物种(E.coli,C.elegans)来说,一个标准的illumina lane得到的数据太多了,仅仅用于测一个样本比较浪费,所以可以多个样本加不同的adapter放在一起测。
数据分析概述
1. 数据分析的概述如下图: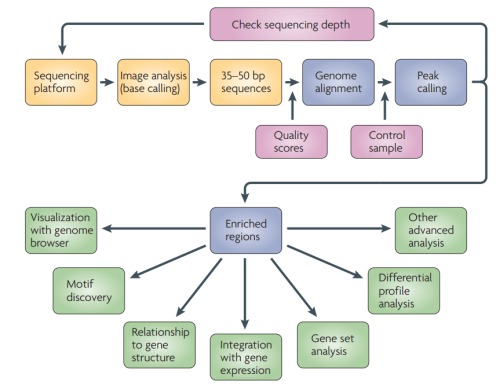
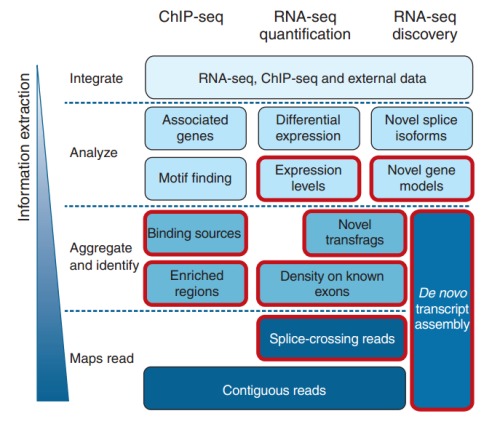
2. ChIP-Seq的主要特征
2.1. “好”数据的特征:
1). 与非特异性的染色体背景相比,从研究目标上得到了足够的DNA片段;
2). 测序文库很全,基本包含了所有的想要研究的片段(是不是测序深度足够的意思呢?)。
“好”数据的数据量:2-20million mapped reads
2.2. Mapped reads是转换成基因组上每个碱基上的reads数,称为tags。
2.3. 信号值高的位点(tags多的区域)并不总是且不是唯一有生物学意义的信号,中等信号被认为更可靠。
2.4. ChIP-Seq的reads的背景分布常常由空白对照经验拟合得到,一些算法也可以根据数据本身而不用controls得到。
3.三种主要数据分布类型
不同类型的蛋白或者组蛋白修饰会得到不同的峰形。下图中给出了常见的。
CTCF: sharp binding sites;
RNA polymerase II: a mixture of shapes;
H3K36me3: medium size broad peaks;
H3K27me3: large domains.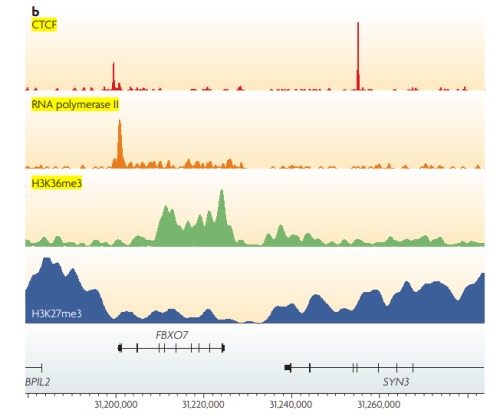
上述峰形的另一种展示是如下(暂且还没有看懂怎么整的):
a. puncate regions covering a few hundred base pairs or less;
b. localized but broader reigons of up to a few kilobases;
c. broad regions up to several hundred kilobases, 大部分有用的组蛋白标记和染色体区域位置.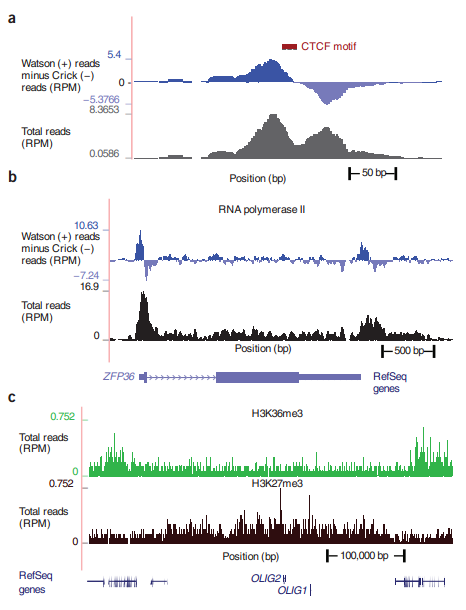
数据分析细节
在数据分析过程中,因为所有和测序相关的步骤可能都会含有reads quality control和mapping,下面直接从ChIP-Seq特有的peak calling 步骤开始阐述。
peak calling
peak finders是指用于ChIP-Seq数据分析的软件包,一般常用来detect染色体区域上的特征峰,peak calling的步骤主要使用peak finders来完成。
一般的peak finders含有5个组成部分(下面的这张图的阐述是很形象的):
1) 染色体上信号波形的定义;
2) 建立背景校正模型;
3) 建立搜索peaks的准则,即建立判断怎样可以是一个peak(一般会用到背景校正模型中的背景值);
4) 校正模型,过滤掉假阳性的peaks;
5) 给找到的peaks排序,给出显著度。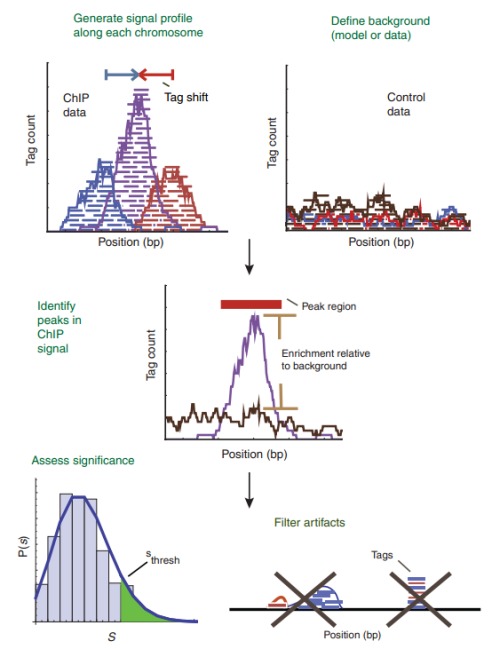
2.1. 探测信号波形(signal profile)的方法(Building a signal profile)
有两种方法:
1.最简单,就是染色体上一段连续的tags超过一个事前定义的阈值就称为找到了一个signal,该方法对于富集度比较大的point source响应性较好,但是中等信号值会被归到噪音或artifacts中;
2. 利用一些额外信息,例如strand-specific(尚未搞懂),如下示例图,定义一个固定的或者动态变化的window width,然后沿着染色体进行扫描。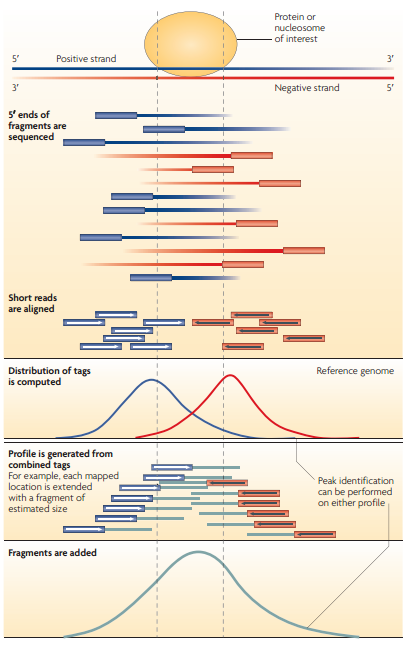
2.2. 对背景值建模(Handling the background)
对背景建模包含假设的统计噪音分布或者一些列的假设用以利用controls来去除treatment里面的背景值、噪音。当没有controls的时候,背景值一般假设使用Possion分布或negative binomial分布来模拟。有controls值的时候一般用treatment的值减去controls的值当做其值。
2.3. peaks 评判标准(peak call criteria)
结合2.1和2.2得到很多候选peaks后,建立统计学检验,查看得到的peaks的显著程度(与随机状况相比)
2.4. 统计后过滤不合格peaks( post-filtering)
一般基于两点来进行过滤:
1. tags between the DNA strands(directionality, 应该就是利用上图中的信息;
2.single-site duplicates(单点重复,应该每个点得有个阈值,少于这个阈值舍去该点) 。
其中directionality的方法中还包括:正反链上的比值,正链减反链的值。
2.5. 显著性排序(significance ranking)
使用p值或者q值进行排序,越小越显著,有些提供FDR.
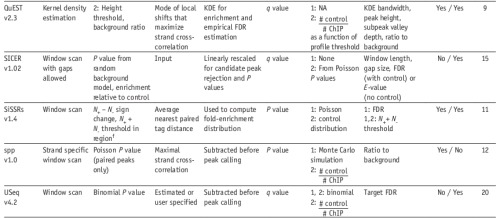
3. 较为常用的软件(09年的review)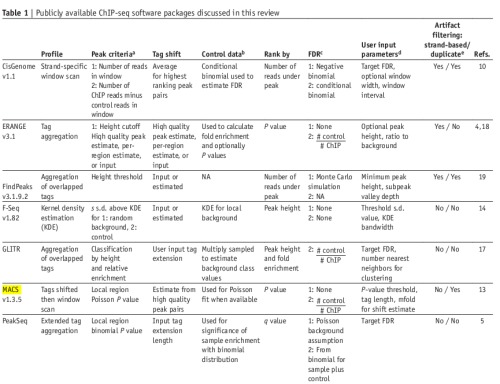
实验与计算术语解释
X-ChIP:首先进行了蛋白质与DNA的crosslink处理,然后进行打断、反交联、测序等等的ChIP-Seq.研究重点是DNA与蛋白质的相互作用。
N-ChIP:实验中并不首先进行crosslink,而是用超声或者MNase直接进行打断,当研究重点是得到核小体的位置和组蛋白修饰的位置。
Multiplexing:对于基因组比较小的物种(E.coli,C.elegans)来说,一个标准的illumina lane得到的数据太多了,仅仅用于测一个样本比较浪费,所以可以多个样本加不同的adapter放在一起测。
tags:Mapped reads转换成基因组上每个碱基上的reads数
region:increased sequence read tag density along the chromosome relative to measured or estimated background.即染色体上的区域出现了上升峰,峰高即为那个对应的碱基上对应的mapped reads数。
source: the most likely source points(s) of cross-linking and inferred binding, 即最有可能的DNA和蛋白质的结合位点。
summit: the local maximum read density in each region. 峰顶,每个region中局部最大峰值。
peak finders: 用于ChIP-Seq数据分析的软件包,应该是指可以detect peaks的工具。
peak calling: 使用peak finders找peaks的分析步骤。
shift: The immunoprecipitated DNA fragments are typically sequenced as single-ended reads, that is, from one of the two strands in the 5’ to 3’ direction. the tags are expected to come on average equally frequently from each strand, thus giving rise to two related distributions of stranded reads. The corresponding individual strand distributions will occur upstream and downstream, shifted from the source point (summit) by half the average sequenced fragment length, which is typically referred to as the ‘shift’. 示例如Fig.1 。 The shift will be smaller and the two strand distributions will come closer together in experiments in which the fragment length, read-length and recognition site length converge.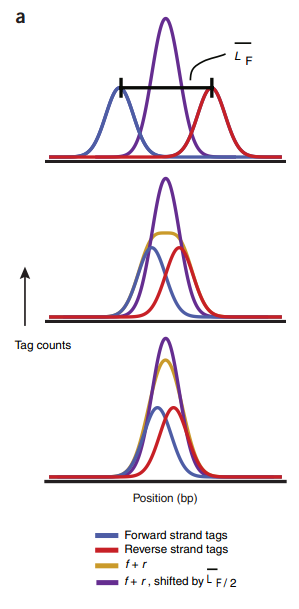
signal profile: 具有较多reads的染色体区域上,将reads数转换成碱基上的tags数,tags的平滑连接即称为signal profile, 这有助于探测区域和更好的summit。
相关文献:
Park, P.J. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet 10, 669-680 (2009).
Pepke, S., Wold, B. & Mortazavi, A. Computation for ChIP-seq and RNA-seq studies. Nat Methods 6, S22-32 (2009).
参考来源:http://caoyaqiang.diandian.com/
- 本文由 ybzhao
- 转载请务必保留本文链接:https://www.plob.org/article/3760.html

若有收获,就点个赞吧
0 人点赞
