基于Hi-C数据的深层挖掘和多组学联合分析已经成为了三维基因组领域的重要组成部分。而工欲善其事必先利其器,夯实基础方能垒砌高台。染色质构象各个层级的识别和鉴定的准确性会直接影响到后续分析的可靠性。去年年底发表在Nat Method的《Comparison of computational methods for Hi-C data analysis》这篇文章详细介绍了十三款软件的优势和劣势。
为了更好地理解后面的内容,小编先介绍下Hi-C文库的原理。细胞核中基因组DNA是一个动态调控的整体,为了能定量地分析染色质在空间上的相互关系,必须将细胞进行固定。甲醛醛基可以是DNA和蛋白中的氨基进行交联,这一过程将染色质构象特征稳定保持在特定的状态中;通过限制性内切酶的酶切活性,使得基因组双链DNA剪切成独立DNA-Protein复合物,即使如此,这个复合物依旧维持在细胞核的原有位置,极少DNA能出核;通过带有生物素的A或C脱氧核苷酸在DNA聚合酶的作用下可以将DNA末端的限制性内切酶粘性末端补平;再通过DNA连接酶活性,可以将平末端的DNA连接成嵌合分子;随后通过超声打断,末端去除生物素和末端补平和加测序接头即可完成Hi-C文库的构建。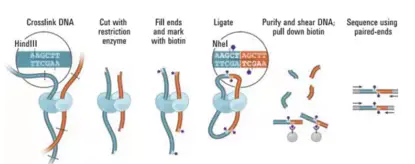
图1.Hi-C实验原理
由此可知Hi-C的标准文库是一个嵌合分子(Chimeric,两侧read来源于不同的基因组片段)。
如果直接将Hi-C的文库拿来比对会发现有效比对率要远低于RNA-SEQ或ChIP-SEQ等文库数据,因为在read两端比对过程中可能存在读过限制性内切酶连接位点的可能。因此在通常情况下,软件一般会选用识别酶切位点预先将reads进行处理或在一端无法唯一比对的情况下,识别连接位点进行切割再次比对的算法(Chimeric mapped)。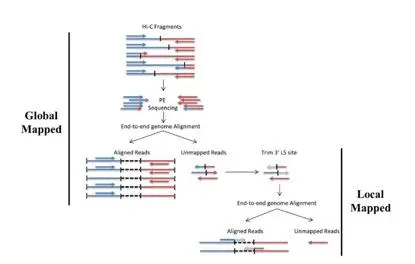
图2.Chimeric mapped算法示意图
言归正传,在这篇文章中作者分析13种用于分析Hi-C数据的算法,它们分别是用于鉴定染色质互作的软件(Fit-HiC,GOTHiC,HOMER,HIPPIE,HiCCUPS,diffHic)和用于鉴定TAD的软件(HiCseg,TADbit,DomainCaller,InsulationScore,Arrowhead,Armatus,TADtree).通过比较发现各种算法之间在鉴定染色质互作中显著的差异和对TAD识别更多可比较性的结果。作者同时选用了已经发表的9个数据集41个样品进行分析,这些样品覆盖了不同的Hi-C实验方法,数据分辨率和细胞类型。
一、比较不同软件在数据比对、过滤的数据产出
Reads 比对。
文章发现相较于Bowtie算法,HIPPIE(采用STAR),HiCCUPS(采用BWA),diffHiC(采用Bowtie2)分别在比对率上提升了18.4%,27.4%,40.1%,同时他们测试利用不同的分析读长来进行比对,发现利用chimeric的方法相较于全长进行比对的算法,能增加30.9%和55.4%的比对数据。表明chimeric在识别更多嵌合片段上更有优势。
无效数据过滤。
在进一步经过过滤步骤之后,测试结果表明,HICCUPS能获取到最多的比对数据,虽然该方法只去除了PCR的重复而没有将其他潜在的无效数据进行过滤;DiffHiC通常会过滤掉最多的比对reads,值得注意的是由于该软件在比对过程中,能得到较高的比对率,所以依然能获得较多的有效reads(表1)。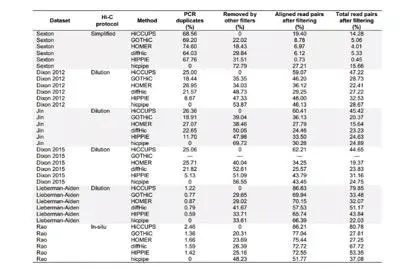
表1.不同数据集中各软件数据预处理结果
比较不同的Hi-C实验方法。发现经过过滤后,in situ HiC能获取到更多有效的reads。对于简化的Hi-C方法则经过过滤后得到最少的reads,通过测试数据发现,这主要是由于PCR 重复率过高导致的(图3c)。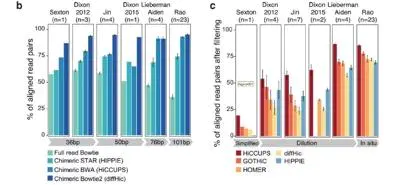
图3.不同软件不同分析读长的唯一比对率及过滤比较
二、染色质相互作用的识别
数据量对鉴定Loop数量影响
研究结果表明,所有方法鉴定得到的顺式互作的数量均多于反式互作,随着有效数据的增加,有效的互作数也随之增加,虽然每个软件的增加有所差别;同时发现染色质内的有效互作要高于染色质间的互作;在大部分的数据集中,GOTHiC能获得最多的染色质内互作;通常情况下,diffHiC软件能鉴定得到最多的染色质间互作,同时发现有效数据量与顺式作用比上反式作用的比例成正比。文章着重提到了HiCCUPS软件,由于该软件采用的局部背景模型,通过图形算法对邻近的peaks聚集成一个单独的互作,因此相较于其他的工具,获得的互作最少(图4a)。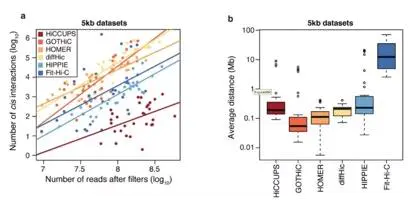
图4.不同软件在不同数据量中鉴定loop的数量和大小分布图
Loop 的大小
通过对顺式互作中染色质内两个bin进行距离分析发现,GOTHiC鉴定得到的互作具有最小的距离,在5kb分辨率下,Fit-Hi-C得到的互作的平均距离要大于10Mb。在1Mb的分辨率条件下,所有软件鉴定得到的互作的平均距离分布在10Mb到53Mb(图4b)。在图中将互作标注在互作矩阵中,可以直观地观测到不同软件鉴定得到的loop差异(图5)。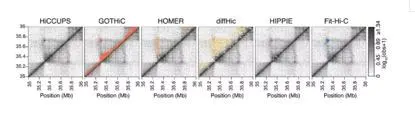
图5.不同软件calling得到的Loops在chr21局部热图中可视化
Loop可重现性
为了比较在不同重复中获得的染色质互作的可重复性,作者选用了Jacard相似系数(两者交集比上两者并集)作为一个鉴定互作集合的度量。一般情况下,相同数据集的技术重复或生物学重复在不同的分辨率中均比较低。然而其显著性要高于随机组合。在低精度时GOTHiC具有最高的一致性,最可能的原因是在每个样品重复中它获得了大量的近距离互作。相反,在几乎所有的高分辨率数据集中,HiCCUPS鉴定得到的loop是各重复中最保守的。令人惊讶的是,与顺式相互作用(JI<0.03)相比,反式相互作用的相似系数反而更高(JI=0.19)(图6d)。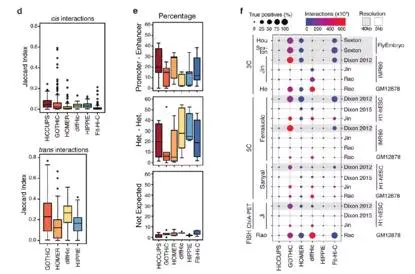
图6.不同软件的可重现性比较特定生物学元件富集
仅考虑top 1000顺式互作的 JI指标,相较于所有的互作的JI,结果发现除了FitHiC之外, 并没有显著改善。相反,当样本中valid pair reads增加时,可重复性也随之增加,HiCCUPS和GOTHiC尤为显著。同时HiCCUPS和GOTHiC软件得到的loop在使用重复相关系数(一个比较不同数量相互作用鲁棒性的相似性度量)来衡量时也呈现最高的重复性(图7a)。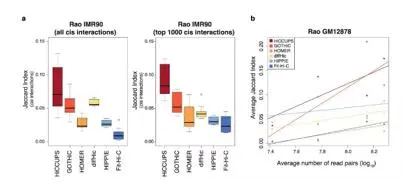
图7.不同软件鉴定的顺式互作的可重现性比较图
在比较同一个细胞系中用不同限制性内切酶进行处理的数据集时,可重复性相似,相反对数据集间之间的重复性(如不同数据集中同一细胞系利用不同的实验方法或酶时),JI系数非常低。
Loop 注释
为了鉴定这些软件calling 得到的互作是否与转录调控的染色质状态有关,作者基于互作两侧的端点基于染色质状态进行分类,在5kb的分辨率考察了所有的方法,发现将近有16%的顺式互作被归类到启动子-增强子,23%互作是分布在异染色质或沉默状态的染色质上。同时3%被认为是缺少生物学支撑,如一端是启动子或增强子,另一侧为异染色质或沉默状态的染色质。
在5kb分辨率下,HiCCUPS和HOMER鉴定得到了最高比例的增强子-启动子的互作元件,虽然他们不是数量最多的(图6e)。在40kb分辨率下,所有的方法均鉴定得到较高比例的增强子-启动子互作,作者认为这可能是较大的划bin更可能包含启动子或增强子。相反,对于反式作用的互作比例,归类为启动子-增强子在所有的软件中均较低。
不考虑软件和分辨率的情况下,少于8%的顺式互作被归类到违背生物学预期的互作中。对于所有的软件,随着数据量的增加,启动子-增强子的占比增加。
Loop 真阳性
当比较鉴定得到的顺式作用数据有效性时,GOTHiC获取了数量最多的真阳性的互作,HOMER和Fit-Hi-C与GOTHiC具有相似的效果,尽管他们鉴定得到的相互作用较少。在高分辨率的数据集中(Rao et,.al),diffHic鉴定的互作具有最高的真阳性比率,虽然在相同的数据条件下HOMER软件比其他所有的软件获得更多的真阳性(图8)。所有的软件在鉴定真阴性上性能均不是很好。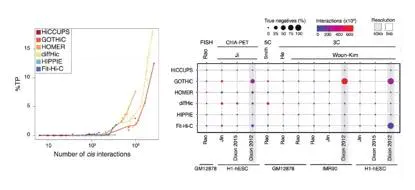
图8.各软件真阳性比例图
三、鉴定拓扑学相关结构域
数据量对鉴定TAD数量的影响
为了进行TAD calling,该项研究用40kb分辨率对所有样本进行分析,与鉴定互作的软件不同,除了Arrowhead以外的TAD 的工具在鉴定TAD时,valid pair数量的增加对TAD的个数影响不大(图9a)。
TAD数量
不同的软件得到的数量不等,在所有数据集中,TADtree软件得到最多的TAD(7638),而Arrowhead识别的最少(636)。相反地,在1Mb分辨率时,InsulationScore能获得最多的TAD。将TAD标记在热图中,可以明显发现HiCseg,TADbit,InsulationScore获得的TAD是连续的,而其他的几款软件则TAD之间有间隔。Arrowhead 和TADtree采用多尺度的方式返回嵌套的TAD。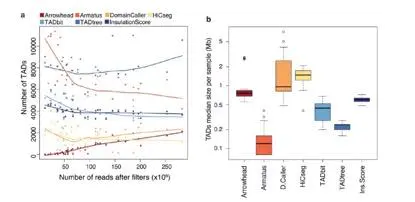
图9.不同数据量calling TAD个数和大小统计
TAD 可重现性
为了比较TAD的可重现性,作者计算了JI作为不同生物学重复间的TAD边界重叠的评价指标。在所有的分辨率下, HiCseg在同一数据集的不同重复中具有最高的可重现性,通常情况下,TAD的可重现性要远高于我们观测到的染色质互作的可重现性(图10)。刚提到测序有效数据的增加不会影响TAD的个数,但是它可以提高所有的TAD鉴定软件在识别不同生物学重复TAD中的可重现性。HiCseg鉴定的TAD在使用重叠相关系数时比其他工具更好。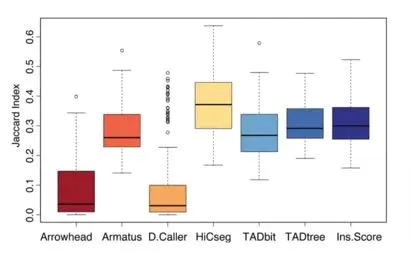
图10.不同软件JI比较
对于相同细胞系而使用不同的限制性内切酶时,用大多数工具得到的重复性是相似的。另外,在不同数据集(同一细胞系不同的实验方案和酶时)大部分软件得到的可重现性要低于同一数据集的结果。TADtree在测试中表现的最好,而Arrowhead表现最差(图11)。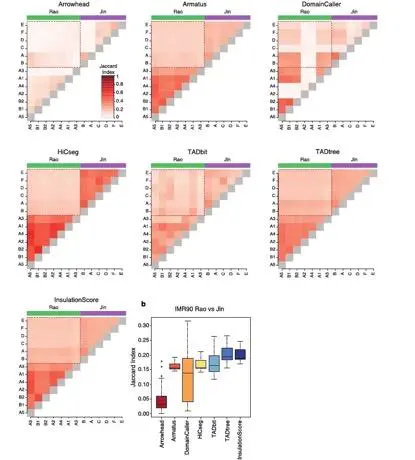
图11.不同软件对不同数据集TADcalling 可重现性比较
TAD 注释
所有软件鉴定得到的TAD在其边界具有绝缘子(CTCF或者BEAF32)的富集。在绝大部分的数据集中超过50%TAD边界都与CTCF的peak重叠。其中Armatus 和 TADtree 获得的TAD边界CTCF的富集尤为明显(图12)。
在Sexton数据集中,大多数软件返回的TAD的边界富集了BEAF32,一个在果蝇中被报道的比CTCF更广泛富集与TAD边界的结构蛋白。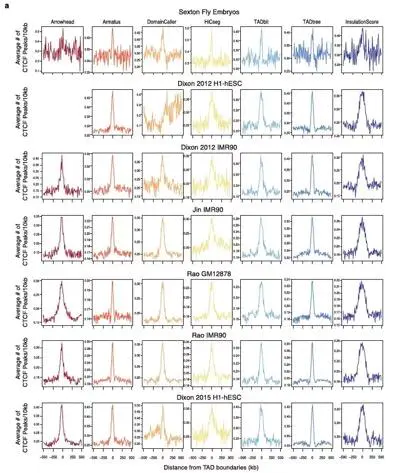
图12.CTCF在不同软件鉴定得到的TAD边界富集图
当使用模拟数据时,DomainCaller,TADbit 和InsulationScore鉴定得到TAD个数与模拟的数相似,且与噪音的增加无关,而HiCseg则鉴定得到少些的范围比较大的TAD,而TADtree则得到更多小的TAD。这两种方法在鉴定正确结构上强烈受到噪音的影响。TADbit和Armatus在重现TAD边界上具有最高的敏感度,尽管TADbit在所有噪音水平上表现的更高精度(图13a)。这一结论同样适用于在模拟嵌套TAD的层次结构时,只不过TADtree被专门设计用于识别嵌套结构域,因此在嵌套TAD的情况中可能会更好些。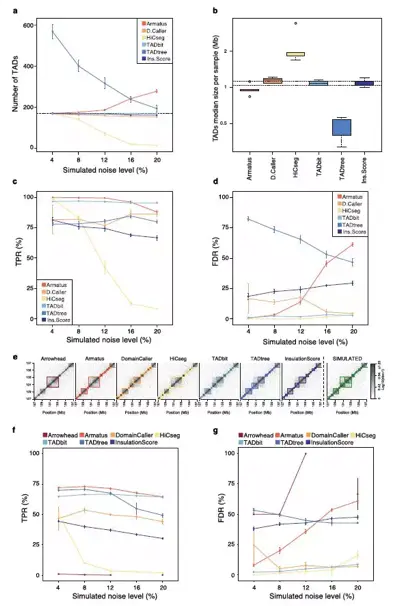
图13.不同软件在模拟数据中表现
四、结论
在讨论部分,作者认为,没有一种算法可被用于作为识别染色质相互作用的金标准。独立于数据分辨率,方法的选择会影响交互数据及其特征。
loop calling方面
- 同一数据中一个技术重复鉴定的loop很难在其他技术重复中重现。而这可能与细胞的异质性决定的。而在高精度的数据中,HiCCUPS具有最高的保守性,在低精度数据中GOTHiC具有最高的可重现性。
- 即使受限于可重现性,所有方法鉴定得到的loop依然是有意义可比较的。loop富集了具有统计学意义的启动子-增强子互作,而只有极少部分的loop是聚类到无法用生物学概念解释的那部分。
- valid pair 数据量增加能增加loop的可重现性和鉴定的loop的数量。
- HOMER和HiCCUPS则能获得更高比例的具有生物学意义的互作,虽然 HiCCUPS的识别潜力仅在非常高分辨率的数据集分析中才能发挥的淋漓尽致。
TAD calling 方面
- 尽管没有一种单独的方法在所有境遇中技压群雄,TAD工具在方法学上较互作的工具要成熟的多,在所有的TAD 识别工具中,TADbit,Armatus和TADtree在实验和模拟数据中的大多数分辨率条件下具有稳定的表现。
- 在实际数据中HiCseg软件具有最高的可重现性,但是在模拟数据中随着噪音增加,其性能显著下降。
- 不同软件calling得到的TAD边界均有结构蛋白的富集。
文章提到在实际数据和模拟的数据中,调和两个之间的差异是很困难的,特别是针对互作的鉴定软件,这可能是由于设计具有预定义特征的HiC数据集的策略复杂性导致的。虽然在对于聚合物折叠建模的生物物理学中具有几种有前景的建模方法,但迄今还未提出任何算法可完全模拟Hi-C数据中的真实分布和偏好性。合成数据的易用性对于合理调整任何算法参数而言都是必不可少的,因此这限制了在选择固有的启发式算法时的最佳选择。
表2.各软件的下载地址
参考文献
Forcato M, Nicoletti C, Pal K, et al. Comparison of computational methods for Hi-C data analysis[J]. Nature Methods, 2017, 14(7):679.
作者:Ray钱
链接:https://www.jianshu.com/p/92fdd30532a5
来源:简书
著作权归作者所有。商业转载请联系作者获得授权,非商业转载请注明出处。

若有收获,就点个赞吧
0 人点赞
