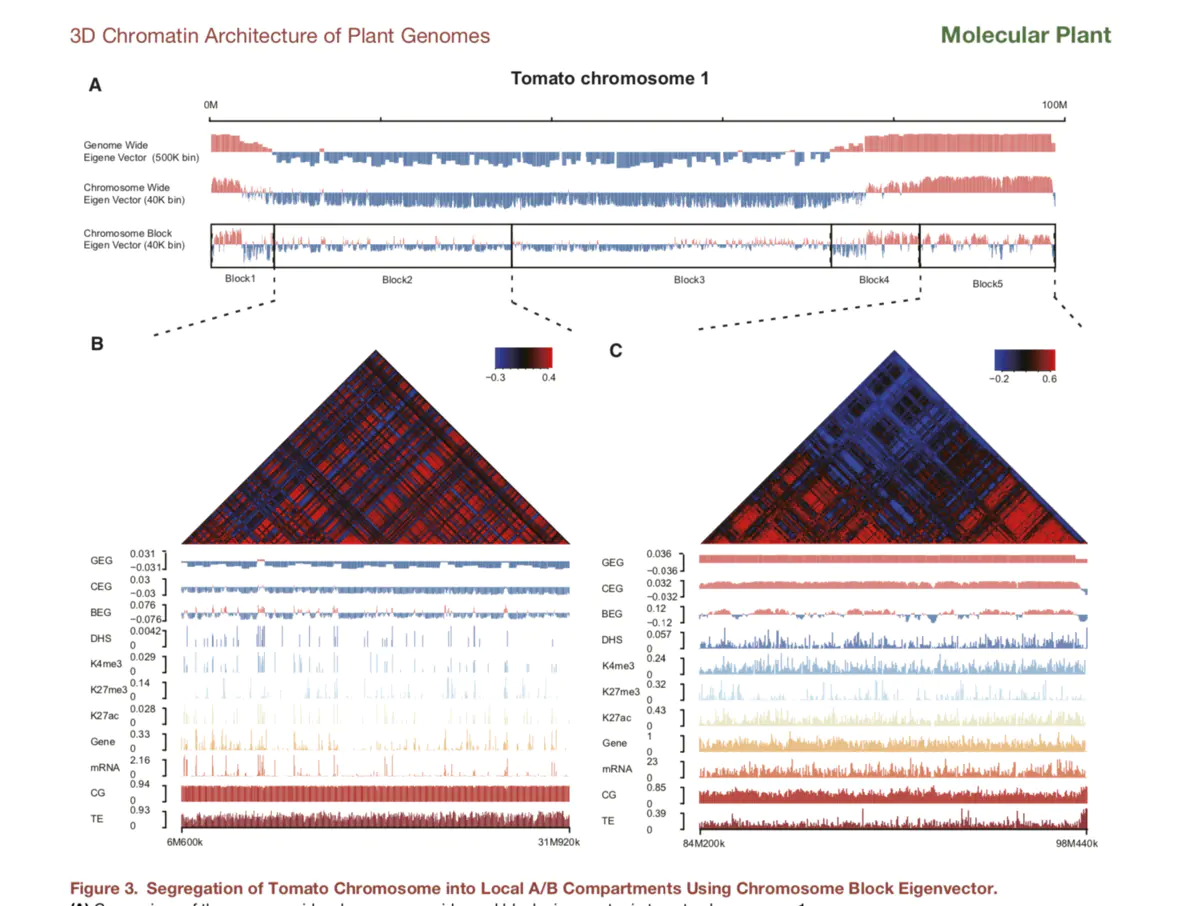

在前面我们介绍了AB compartment分析的软件 cworld 软件(https://www.jianshu.com/p/2cd156681a46),但是cworld 软件是用perl+python组合来写的,计算效率不高,需要内存大,这就导致了我们在进行高分辨率AB compartment 分析的时候,往往会遇到这样的情况——程序运行了很久,最后报了一个“Memory Error”之后销声匿迹。
针对这个问题,今天推荐两款软件:
一、juicer 软件:
juicer 软件能够以5kb的分辨率来计算eigenvector (如下图)。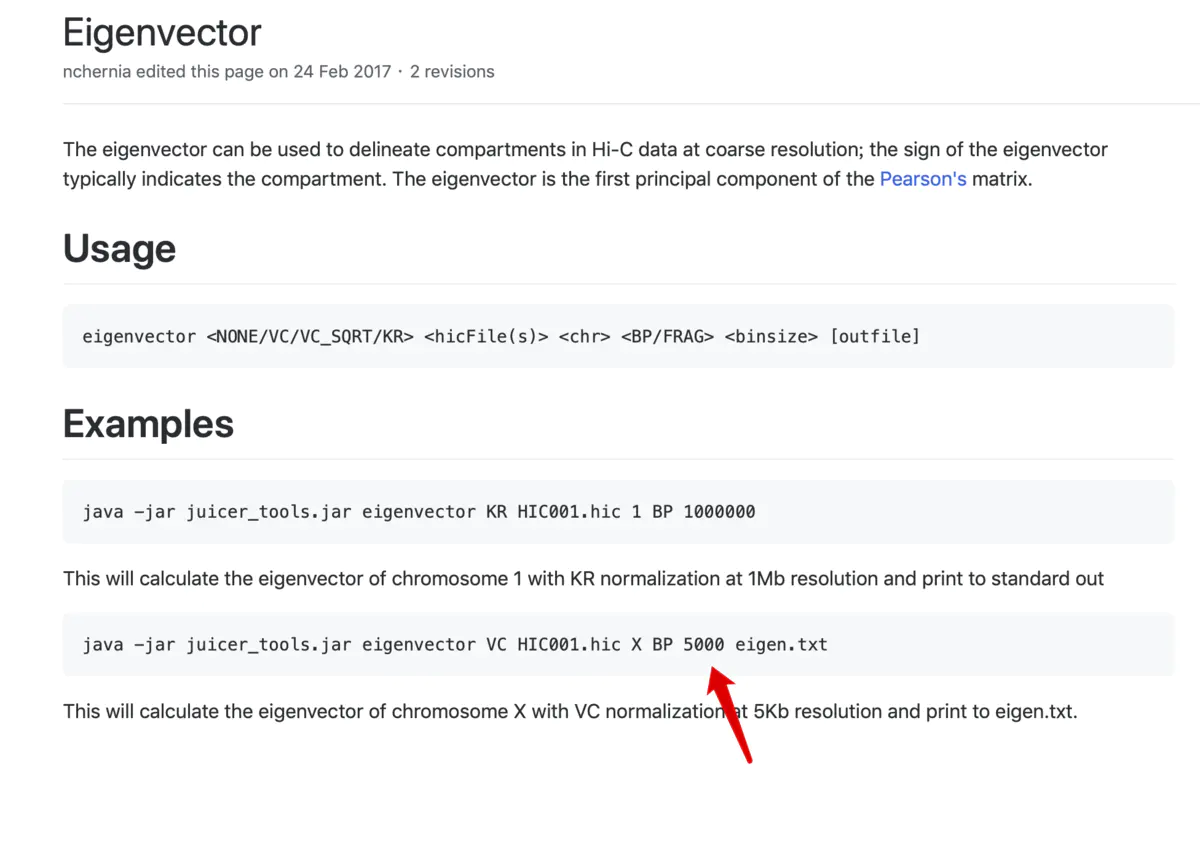
二、CscoreTool (https://github.com/scoutzxb/CscoreTool)是一款分析compartment的快速工具。
Usage: CscoreTool1.1 < windows.bed> < input.summary> < outputPrefix> < session> < minDis> [chrName]
输入参数:
a. windows.bed—此文件用于指定要分析的基因组窗口。它应该是等长的bed文件,覆盖目标区域,大概是整个染色体或整个基因组。
windows.bed 文件格式:chr1 1 10000chr1 10001 20000chr1 20001 30000chr1 30001 40000chr1 40001 50000chr1 50001 60000chr1 60001 70000chr1 70001 80000
b. input.summary—该文件是Hi-C交互的主要输入文件(HicPro 生成的validpairs文件前7列)。
vaildpair 前7列:D258GACXX130605:6:1106:12664:6611 chr1 755519 + chr1 755925 - D258GACXX130605:6:2114:9811:10334 chr1 808619 + chr1 175784252 - C24LCACXX130513:6:2205:5035:47173 chr1 841140 + chr1 801416 - C24LCACXX130513:6:1203:16707:42415 chr1 853901 + chr1 880189 - D258GACXX130605:6:1116:10492:97284 chr1 855471 + chr1 855853 - C24LCACXX130513:6:2305:3295:12387 chr1 869064 + chr1 838284 - D258GACXX130605:6:2215:16114:89299 chr1 893617 + chr1 894051 - C24LCACXX130513:6:2204:16005:64911 chr1 910016 - chr1 5806178 - C24LCACXX130513:6:1214:10668:2084 chr1 929444 + chr1 940247 + D258GACXX130605:6:1101:5936:36199 chr1 931624 - chr1 950942 -c. OutputPrefix—这是输出文件的前缀。
- d. session—这是要使用的会话数。会话的数量取决于可用的资源。该程序未完全并行化,因此使用更多会话只能部分提高速度。
- e.minDis—这是要考虑的最小交互距离的标准。它应不小于分析分辨率。我们建议使用1000000(1M),因为与A / B间隔相比,更短距离的交互可能受TAD结构的影响更大。
- f. chrName—这是一个可选参数。用户可以指定某个染色体进行分析。
示例运行:
CscoreTool1.1 hg19_chr1_10k.bed Test.summary Test_chr1_10k 12 1000000 chr1
有4个输出文件。
- XXX_bias.txt—这是每个基因组窗口的估计偏倚因子。
- XXX_hh.txt—这是估算的距离曲线。相应的距离是10 ^(0.04 * n),其中n是第一列中的数字
- XXX_cscore.txt—这是每个基因组窗口估计的Cscore
- XXX_cscore.bedgraph—这是用于可视化的基础图文件。低映射窗口(偏差<0.2)或高拷贝数窗口(偏差>5)已被过滤。
CscoreTool论文中涉及分析的所有基准图文件都可以在Bedgraphs.zip文件中找到。
核心结果文件XXX_cscore.bedgraph:
track type="bedGraph" name="test_cscore.bedgraph"
chr1 710000 720000 -0.203475
chr1 750000 760000 -0.857018
chr1 760000 770000 -0.9999
chr1 770000 780000 -0.9999
chr1 780000 790000 -0.703065
chr1 790000 800000 0.366704
chr1 800000 810000 -0.9999
chr1 810000 820000 -0.9999
chr1 820000 830000 -0.9999
作者:XuningFan
链接:https://www.jianshu.com/p/0e860a6b3036
来源:简书
著作权归作者所有。商业转载请联系作者获得授权,非商业转载请注明出处。

若有收获,就点个赞吧
0 人点赞
