写在前面
TBtools开发至今三年有余,让开发工作的持续开展,事实上主要来源于用户朋友的信任与支持。总的来说,我对多数用户存在某种意义上的感谢,毕竟使用一个未正式发表的工具,并在漂亮的发表工作引用,是对我个人能力在某种程度的认可。
与开发早期极少数负面评价相同,同样存在一部分非常支持TBtools的开发工作。部分朋友,甚至将TBtools应用于学校课堂教学。这是我从未敢想的事,毕竟TBtools事实上只是我们课题组(前面硕士与现在博士)分析需求的部分体现。
我是一个潮汕人,潮汕人(或者可能是我家人给我的感觉)有一个传统,即知恩图报,更或者说,滴水之恩,涌泉相报。
开发这一功能的起因
在非常支持TBtools开发的用户朋友中,其常常与我提起,
能否开发出一个支持正则表达式检索的工具?用于查找结构域或者特定的模式,如微卫星….
总的来说,我觉得这个功能似乎我并不一定用得到,所以我或者会建议其去找另外的工具,比如MEME suite的MAST,或者自己编程实现。但是最后,似乎他还是没有按照我说的去操作。
昨晚我在等待数据下载的过程中,再次看到其提起。我想这已经是其超过三次的提起,或许确实可以花时间试试,毕竟辜负一个一直支持你的人的期待,似乎是一种不讲义气的操作。我大体想了下,这个事情似乎还不是我以前想的那么简单。
- MAST是不允许gaps的,其最大的作用是允许misM
- 自己变成时,那么多行的序列的回车需要处理;而如果一次将整条序列读入内存,那么遇到chromosome时,可能会有内存不足。
- 序列模式的Overlap的情况需要处理
- 不定长度的正则要求,可能确实不好找到工具来处理
总的来说,似乎确实找不到合适的工具可以很好的支持这些需求。所以,我写了一个。
功能的使用
首先是打开TBtools,找到最新的功能,Fasta Sequence Pattern Locator
需要设置的是三个参数: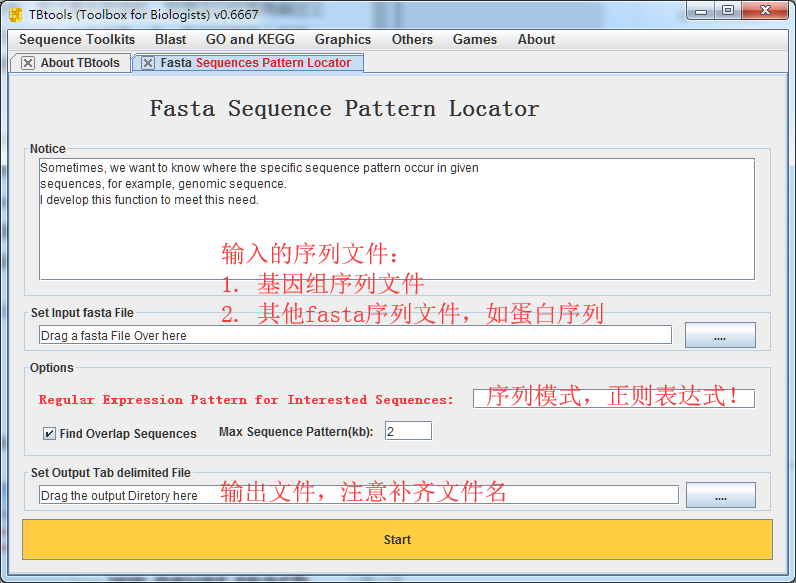
对于输入的序列文件,事实上,需要保证的只是Fasta格式,而与其序列长度与类型(全基因组序列或者蛋白序列等)没有关系。
对于序列模式,那么需要用户对正则表达式有一定的了解,比如挖掘微卫星(AT){6,}表示ATATATATATAT….;或者从mRNA中预测ORF,ATG(?:\w{3})+T(?:AG|AA|GA),随后对每个序列取最长的一个最长ORF即是。当然,也可以使蛋白的某些序列模式…
如果确实不了解正则表达式,其实还是比较简单,大概可能知道
ATCG 对应的就是 四个碱基,ATCG[ATCG] 对应的是 一个碱基,A或T或C或G[^AT] 对应的是 一个碱基,但不会是A或者T(AT) 对应的是 两个碱基,把AT定位一个单元(AT){6} 对应的是 2x6,一共 12 个碱基,也就是AT重复正好6次,如ATATATATATAT(AT){,6} 对应的是(AT)重复不多于6次,如AT, ATAT, ATATAT, ATATATAT, ATATATATAT, ATATATATATAT### 之前有笔误,谢谢“微信-潜”,20191018(AT){6,} 对应的是(AT)重复不低于6次, ATATATATATAT, ATATATATATATATATATATATAT.....等等....
Overlap参数,大体对应的就是模式之间是否允许Overlap, 比如ATG\w{111}TGA,那么在这个模式捕获出来的序列中间是否有可能出现新的ATG?
Max Sequence Pattern(kb),查找出来的片段大小,不能长于这个长度,如2即2kb。这个参数事实上,也对模式查找存在一定的影响。对于Overlap模式的,没有影响。对于不Overlap的,可能会丢失一小部分模式。
当所有参数设置好之后,设置输出文件,注意补齐文件名。随后点击Start即可。
输出结果
输出的结果,主要分为四列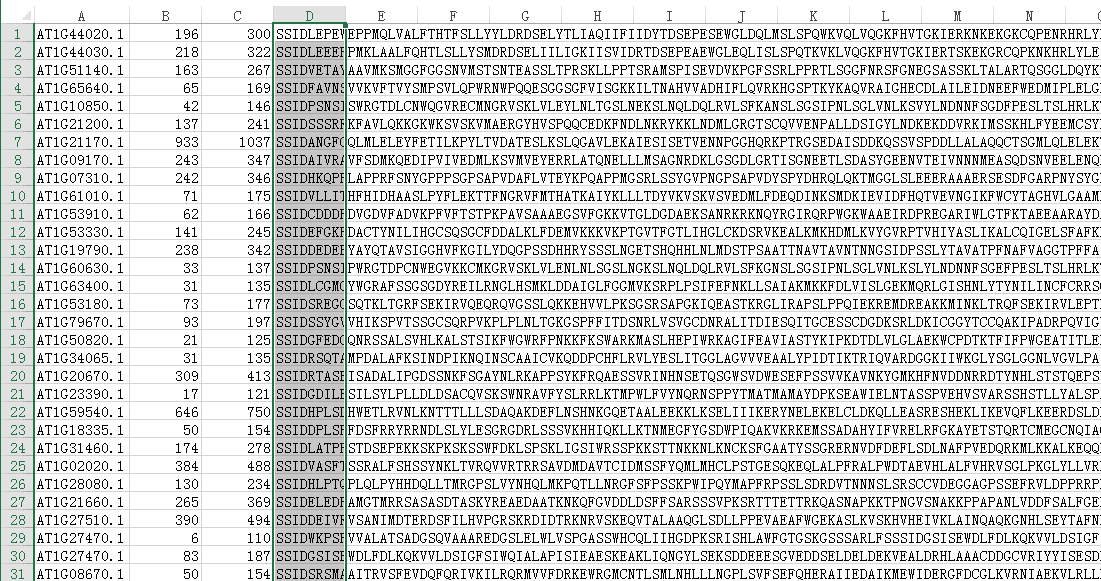
分别是:
- 序列ID
- 起始坐标
- 终止坐标
- 匹配到模式的序列
其实是可以支持多个模式一次查找的,但我确实没有感觉到这个需求的大小,所以暂时也懒得支持。
做一个有趣的事情( 注意,后续在朋友的提醒下,这个操作不适合预测着色粒)
既然,我们写了这么一个功能,那么我突然想到可以拿来做有趣的事情。有关注公众号历史推文的朋友,应该了解到,我们课题组与福建农张积森老师课题组存在较多的交流与合作。我参与了博士导师与张老师的众多合作项目中某物种基因组分析的学习。在他们交流的过程中,我了解到,在张老师发表的甘蔗基因组文章中,AAACCT重复序列可用于预测着丝粒位置。(20190503,发表后一周内,收到朋友提醒,AAACCT似乎应该是端粒的motif….)
于是,我们可以做一下试试。
第一步,挖掘出拟南芥基因组中,所有AAACCT位点位置(我发现,直接设置10个重复位置,直接没有分析结果)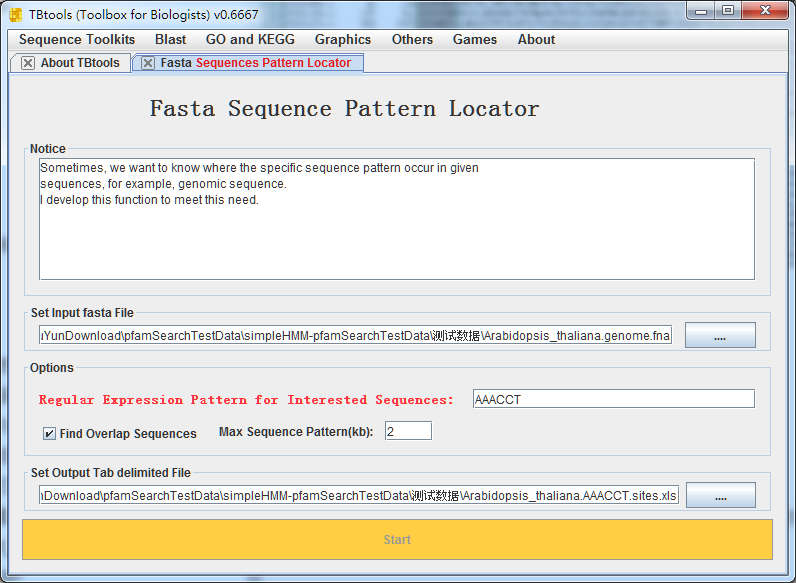
第二步,生成染色体长度文件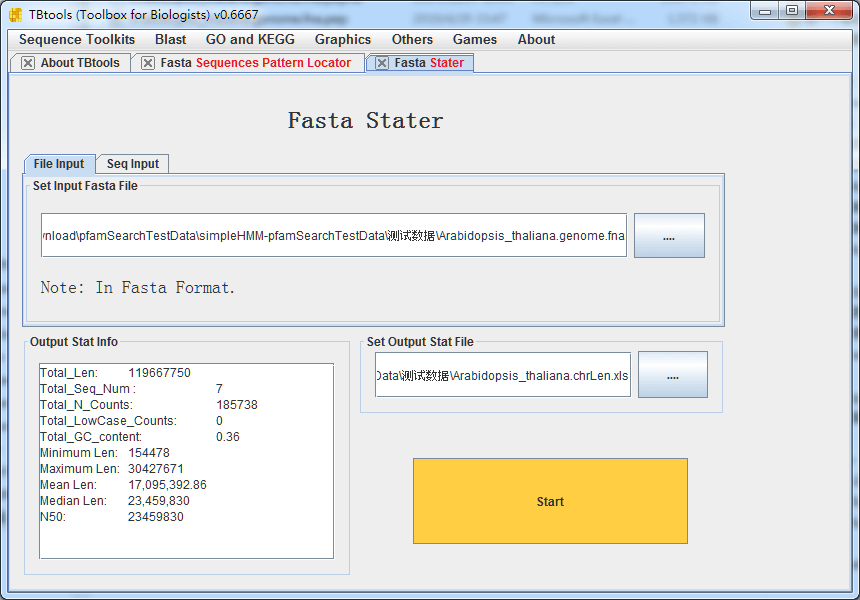
打开并做以下调整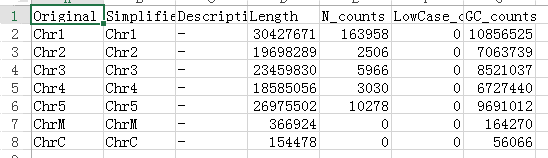
变成以下并保存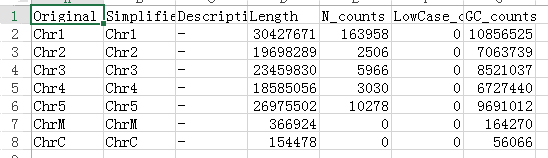
第三步,打开并整理AAACCT模式的文件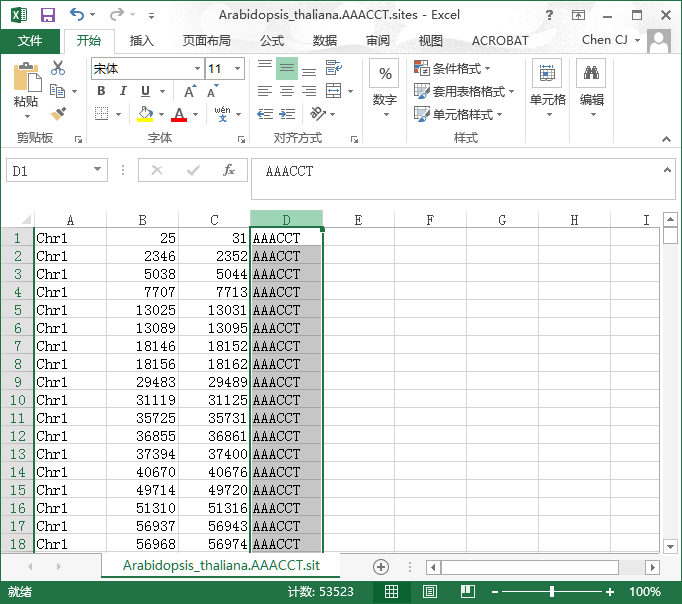
替换第四列为,全部为1
并保存文件
第四步,使用Amazing Super Circos,进行可视化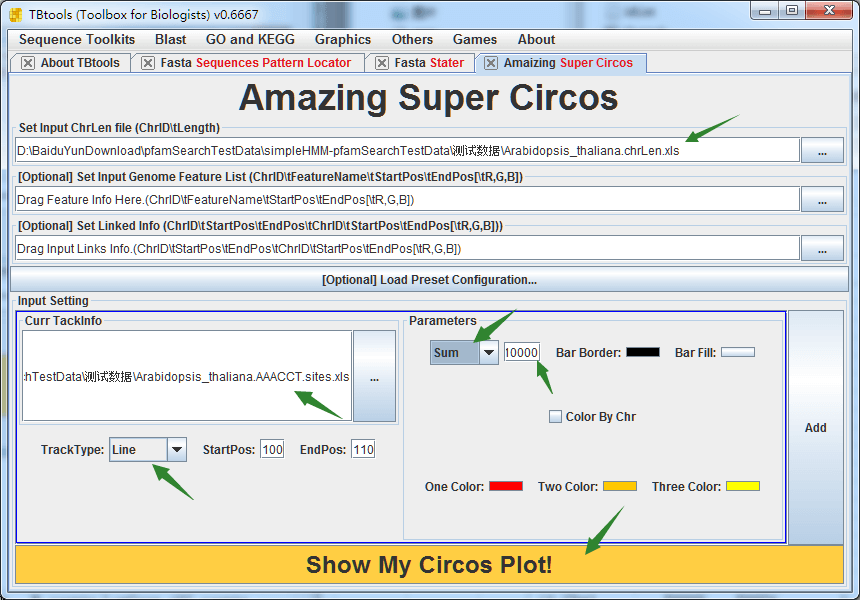
似乎只有两个染色体能够很好的看到峰值,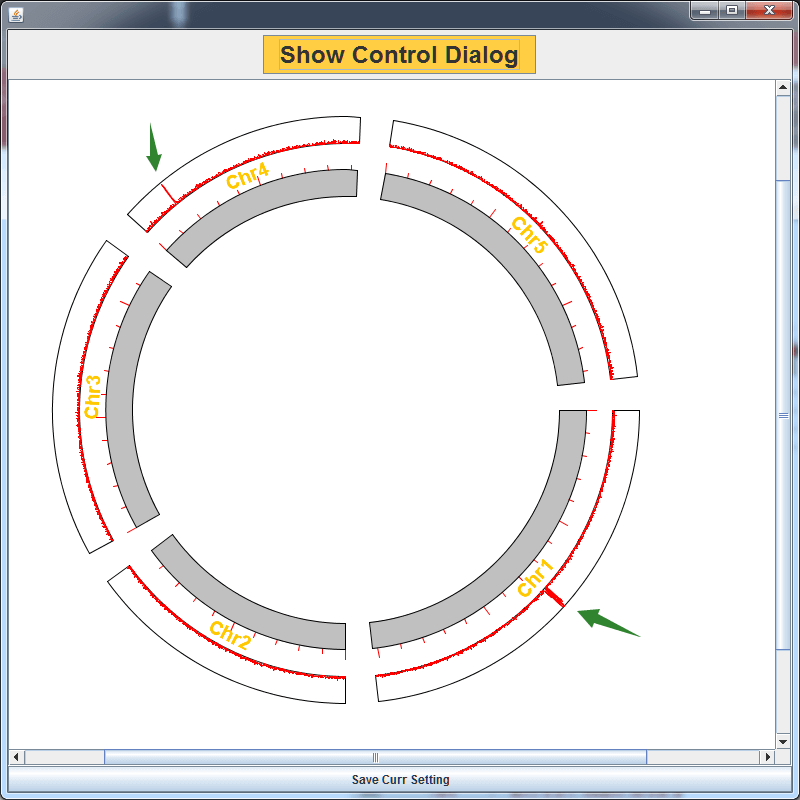
对比拟南芥基因组文章图片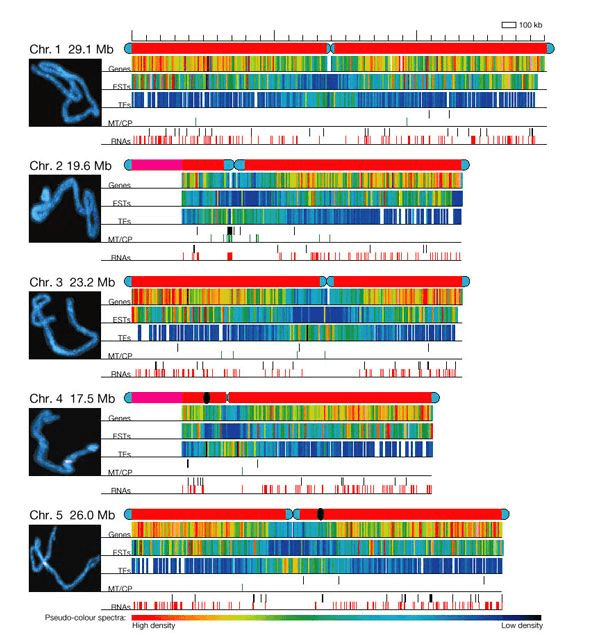
可以看到,似乎Chr1和Chr4基本可以对应上。考虑到,可能是Circos Binning时窗口大小的问题,做微调,设置窗口为1kb,似乎没有太大变化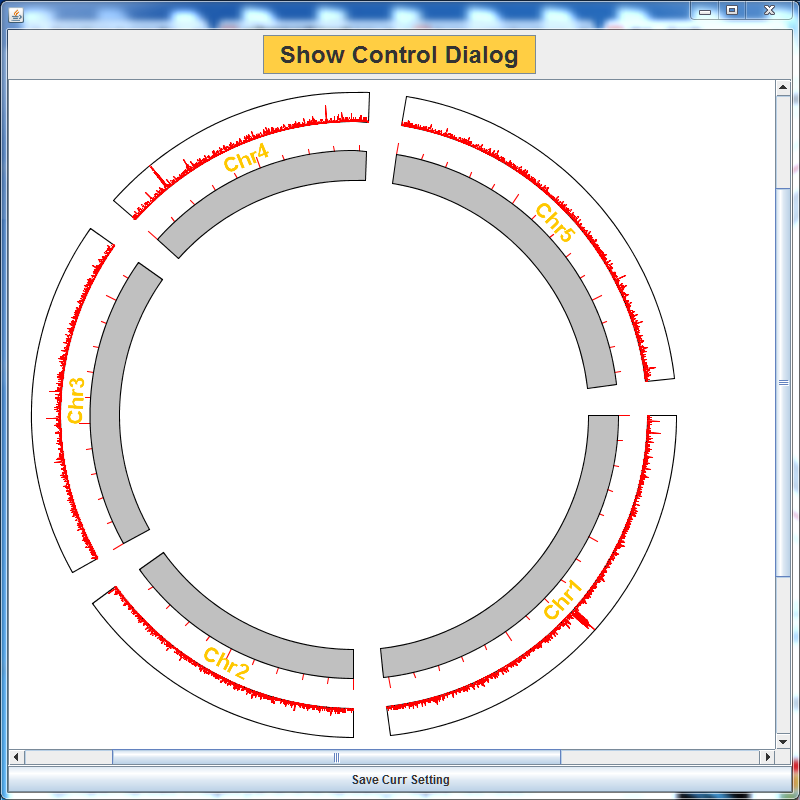
换成我们手上在做的基因组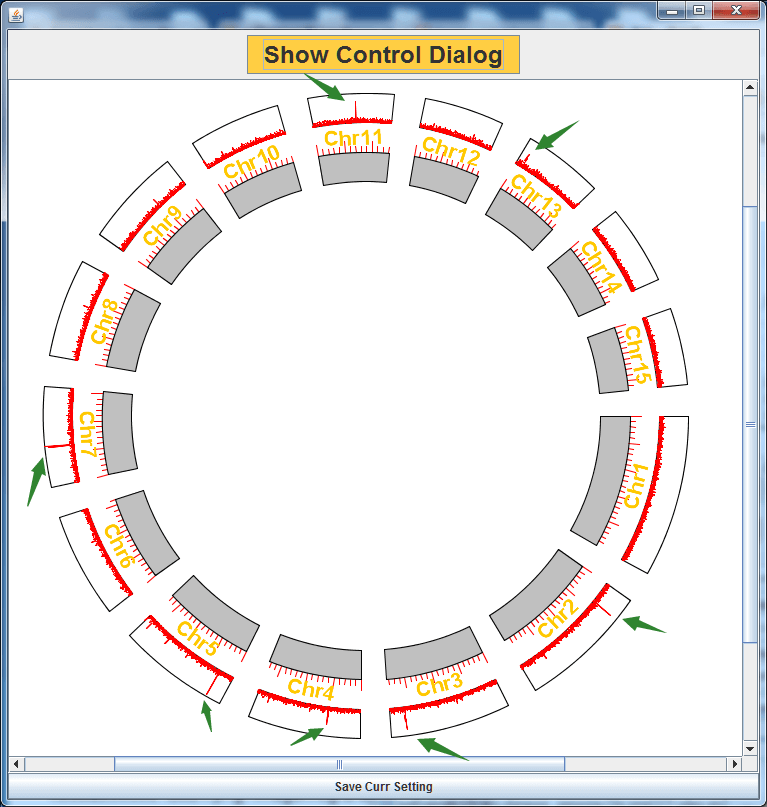
恩,看起来,似乎效果更好一些。只是,这些位置是不是着丝粒位置?
顺手加一圈基因密度图….恩…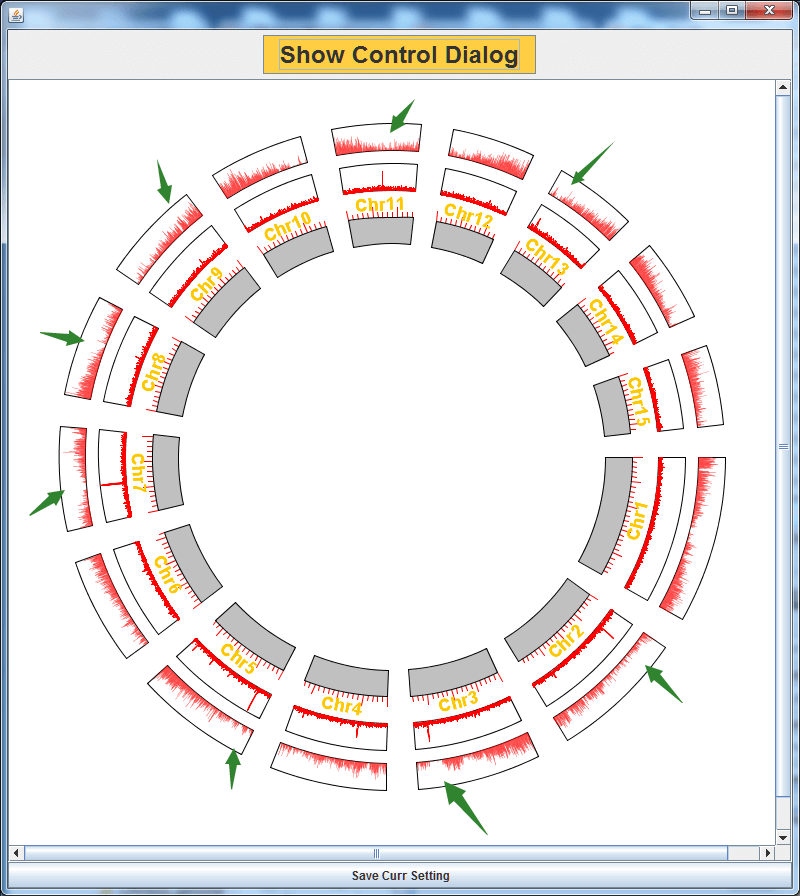
好像可能有点用。不过AAACCT重复的,自然基因也会少,这个可能也不一定是着丝粒位置。
注意,据个人认知,不同物种着丝粒区域的序列重复模式不同。
写在最后
每个人,总是要越活越明白。时间与精力是有限的,所以慢慢的目标就会变得明确。两个事情是需要兼顾的:
- 做好手上在做的
- 不断提升自己的能力

若有收获,就点个赞吧
0 人点赞
