Motif,模式,pattern,如下。我个人理解为:一组具有类似特征的序列。而在分子序列中,那么就是具有类似分子(如碱基或氨基酸)的序列。
Motif,不是一个序列,所以在生物序列分析时,无论是预测和挖掘,都并不是简单的完全匹配就能完成,他的处理,有点像正则表达式,但事实上,应是HMM。这种情况下,使用已有工具,其实是最合适的。
MEME suite是一个motif挖掘和分析工具。在大多数情况下,我们会使用它从批量数据中鉴定出某些保守的motif,比如Chip-seq;当然,我们也会使用某个特定的motif,去海量的序列中,查找可能包含该motif的序列,比如转录因子结合位点或特定元件。
写在前面
Emm……生物信息,应是一门完整的学科。而整个市场在批量生产生信工程师,这个跟IT是类似的,比如批量生产程序员。这种情况的出现,可能是因为存在一些无法自动化的东西,而需要人工干预。这个话题,已经很久没有再谈及。故,这里也不做展开。
一些思考
无论生物信息学怎么发展,但是生物(或者美其名曰,生物信息)数据分析,总是可以有相对简单地方式去完成一些相对简单的目的。我一直对外推荐,新手用python,然而我还是在用perl。推荐python,主要原因是,主流如此。而我还在用perl,是因为我喜欢我所理解的和接受的perl的设计理念。
使简单的事情,可以非常方便地完成;而对于复杂的事情,努力一下,也可以完成。
我最喜欢的是前半句。我之所以这么说,可以看我QQ空间几年前写的perl one-liner日志,里面是一些我现在不太看得上的,但是可以完成很多事情的,单行命令。
如下,单行进行序列批量提取
或者单行获取所有序列长度
perl -0076 -ane '@F=map{s/[>\r\n]//gr}@F;$id=shift @F;print $id,qq{\t},length (join q{},@F),qq{\n} if $id' in.fa
事实上,或许你没有意识过来,上述的perl单行,其实是跨平台的且随时可以copy-paste运行,而你几乎不需要下载任何程序。换句话说,找个linux或者windows(当然perl解释器先安装好),就可以直接黏贴运行,并输出结果
这些想法,出发点,从一开始就长在我的潜意识中,并蔓延到所有我写的脚本或者工具。
很明显,TBtools就是这样一个体现。而他的开发,却被一部分人误解(我其实甚至不知道误解我个人出发点的那部分人是否有自己对生物信息的认知)。TBtools的存在,名义上是为了湿实验工作者开发,实际上则是为了在是实验室工作的干实验工作者开发。每一个课题组,每个人都有擅长的事情;通力合作,各取所长,才是整体前行的最大助力。作为一个自称搞分析的,难道你最擅长的是帮别人提序列?做热图?做Venn?跑本地Blast?….,我想,不是吧。以任何方式,消灭一些高重复低复杂的工作,才是事实上节约彼此时间的事情。
Windows下直接运行MEME suite
课题组最近做了一些测序,需要做类似的分析。我个人的建议是,把序列抓出来,然后到MEME suite官网跑一下看看。结果是什么?无尽地等待!MEME suite网页分析常常需要排队,且每次排队可能是一两天。解决的办法只有两个:
- 上服务器,命令行运行
- 让其他人上服务器,命令行运行
而我变成了上面的其他人。分析的要求,有各种各样,我有事的时候,其实并不想过多地沟通,但拒绝合作,并不是课题组发展的最好选择。
于是,我小修了MEME和MAST的源码,并编译,随后打包到了TBtools。
于是,所有人都可以自己跑了,即是你在windows下。
使用MEME,任何平台都可以
注:如果是linux或者Mac,需要自行安装好MEME suite….;如果是windows用户,直接TBtools.exe安装器安装即可
使用的示例数据输入为拟南芥的126个MYB蛋白(注意,核酸序列也可以),运行时间大体是2min,序列越多,时间则指数型增长。
与运行网页工具的操作是类似的,设置好对应的参数即可,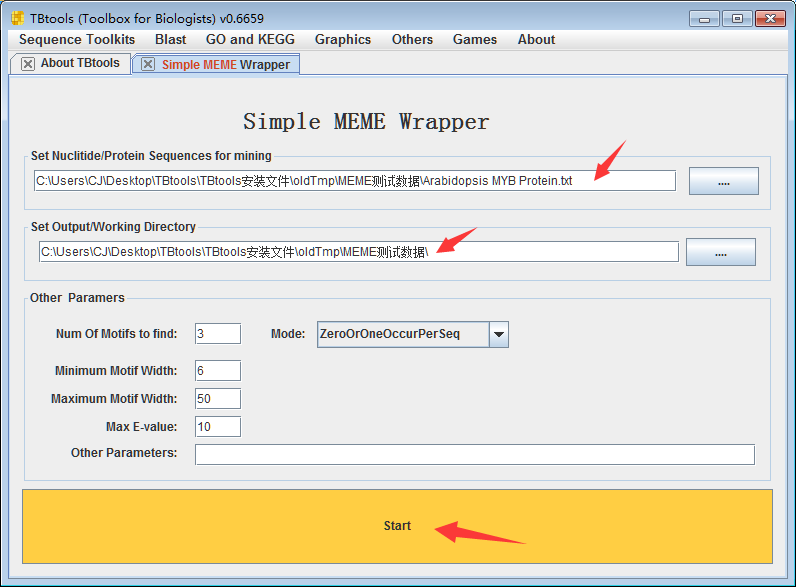
以上,设置motifs个数为3,motifs长度为6到50,得到输出文件如下
其中meme.txt文件,直接用文本编辑器打开,即可看到类似MEME suite网页版运行结果的纯文字版本;
而meme.xml文件可以直接用于TBtools做可视化
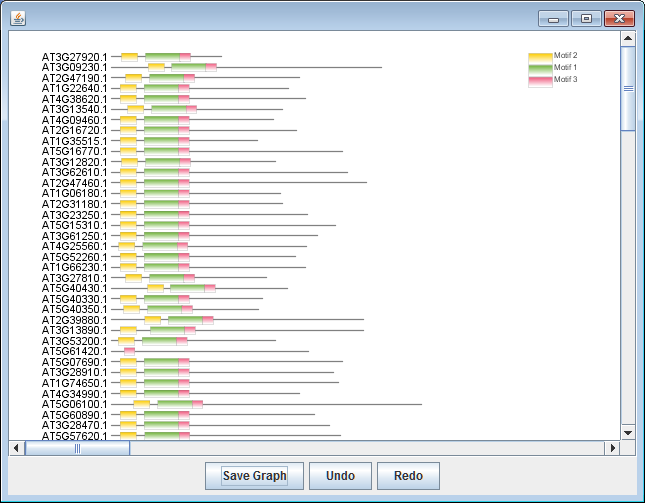
同时,也会得到3个motifs对应的SeqLogo信息
使用矢量图编辑器打开即可
MAST的使用是类似的。当我们获得一些motifs之后,我们可以用这些motifs,从海量数据中快速搜索包含该motifs的序列。MAST的运行是很快的。上万个序列,也是秒级出结果。我们可以直接上述使用meme.xml文件作为输入。这里我们使用原来的蛋白序列集合作为输入。
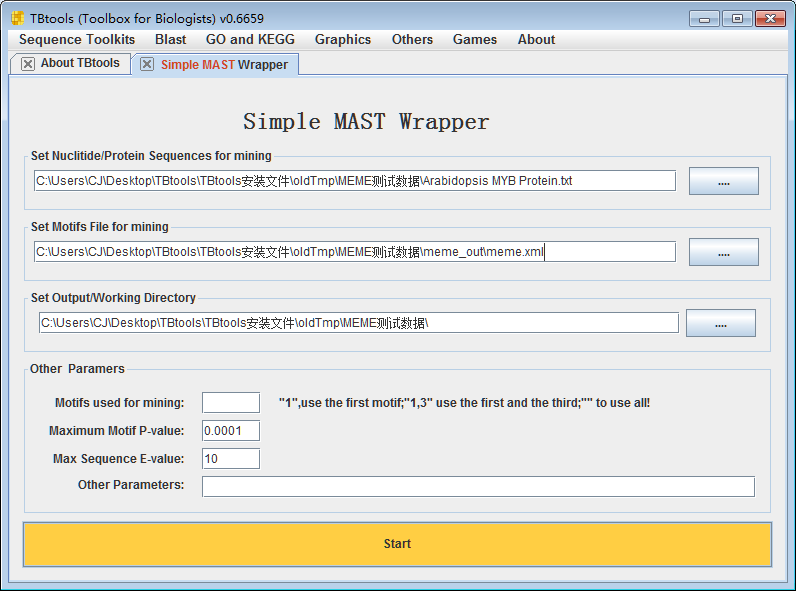
注:MEME的主要目的是从一堆序列中抓出可能的保守模式,在于挖掘;MAST的主要目的是基于模式找位点,在于鉴定。相对而言,后者敏感度可调整性高些。具体使用者,应有自己的理解。
写在后面
但行好事,莫问前程。是吗?

若有收获,就点个赞吧
0 人点赞
