写在前面
目前,有大量物种的基因组序列和注释信息公布。几乎所有科研人员都可以下载并使用这些数据,分析和验证自己的科研假设。常常,我们会遇到一些小问题,比如基因组序列文件中包含了大量无法锚定到染色体的片段。这些片段,当然是有存在意义。但在下游数据应用上,基于不同分析目的,其实并不一定要用上。更或者保留这些序列,反而不便于下游数据分析。举两个例子。
图中菠萝基因组序列文件中有大量scaffold,使得25个染色体(尽管是最大连锁群)无法被很好的可视化,拿到这个图片,自然无从分析。当然,拟南芥基因组序列也是类似,叶绿体和线粒体基因组在这个分析中并无必要存在。
再看,葡萄比对到自身的基因点阵图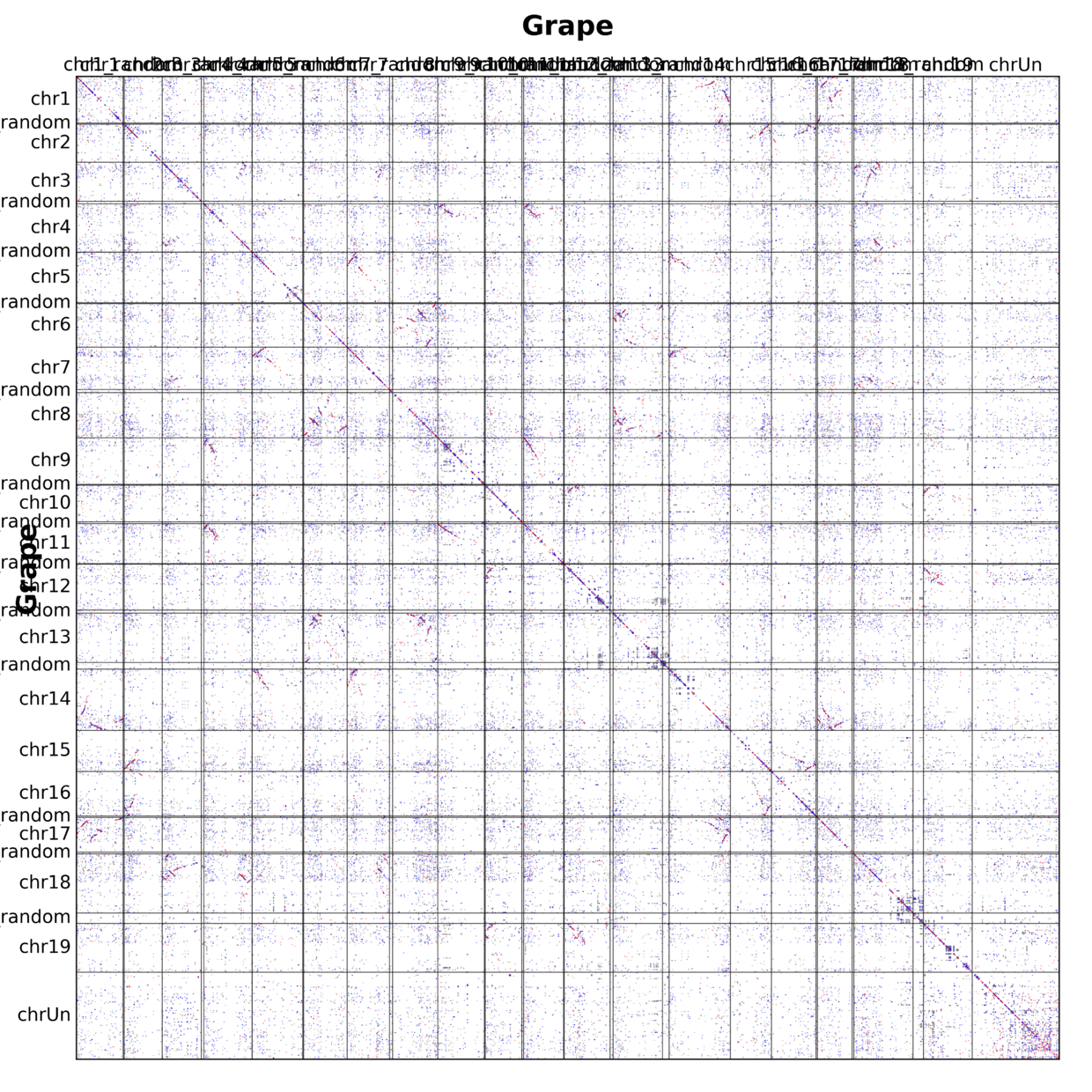
去掉 random 染色体碎片之后,如下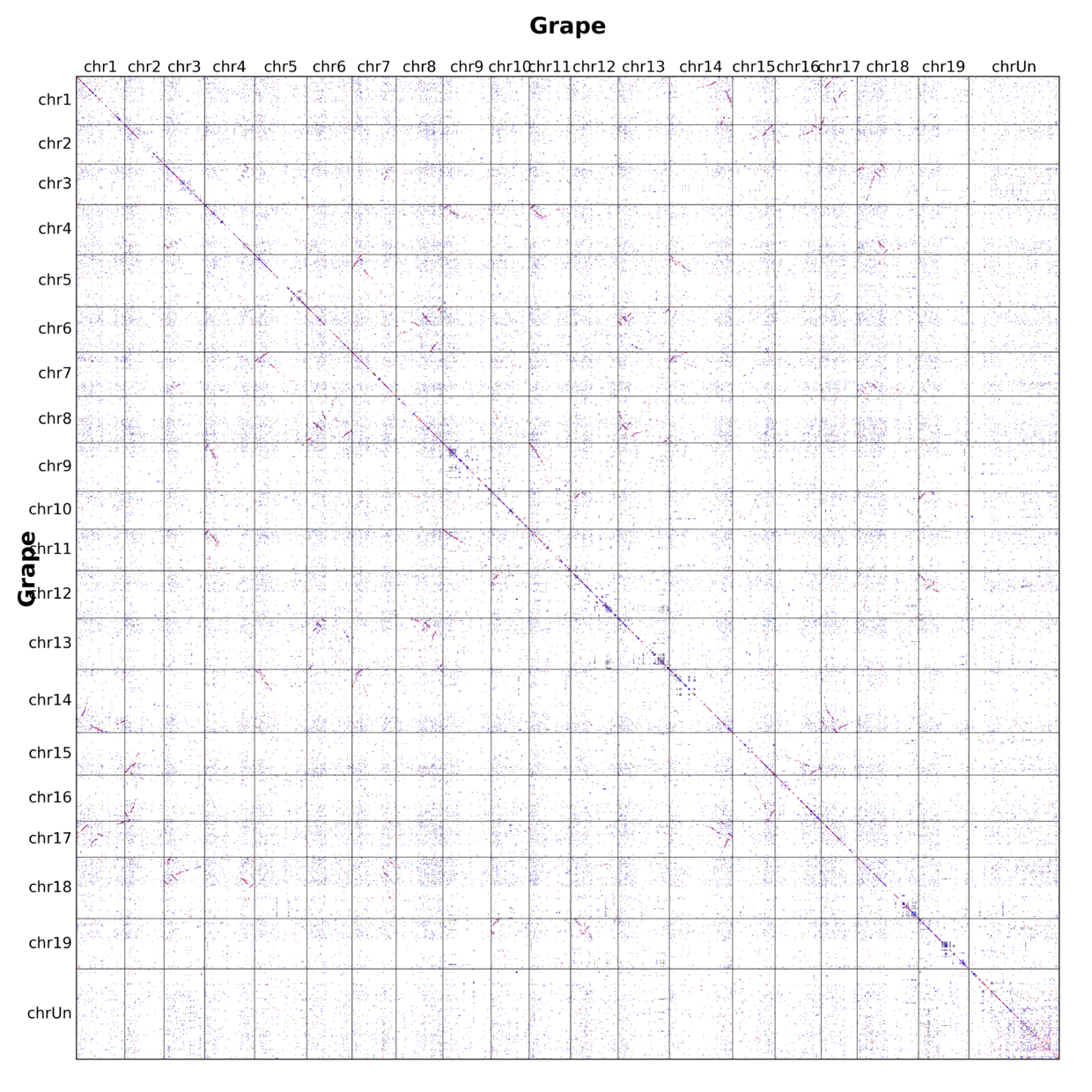
可以看出来,其实图片干净了许多,也才是我们关注的分析内容。
总而言之,过滤掉这些基因组序列中的碎片,常常有助于下游分析。而过滤策略其实往往简单。碎片,自然是长度比非碎片的小得多。所以,我们只需要基于长度做过滤即可。如果使用 之前版本的TBtools ,大体可以通过以下步骤实现:
- 使用
Fasta Stat获得所有序列长度 - 使用 Excel 或者 TBtools 表格工具,对长度做排序,找到合适的长度阈值
- 过滤并保留所有长度不低于该阈值的序列 ID
- 使用 TBtools 提取这些序列
大体有至少换TBtools三个功能和四步操作。当然,一般保险起见,我们过滤完基因组序列,最好还要过滤一下基因结构注释文件.gff3/.gtf。这一步,使用 TBtools 的表格工具,其实也可以解决。
说了这么多,其实只有一句话:太麻烦了!
新功能一次搞定
事实上,我自己也被这个折磨了一段时间。想着有时间就写一点,期间不断被各类事情打断,一个原本可以用大半个小时写完的功能,却整整跨越了一个星期。看了下,感觉确实是一天写三四行代码。当然,这并不影响功能的实现,以及这个功能用起来到底有多爽。
打开 TBtools 并找到这个功能,Genome Length Filter。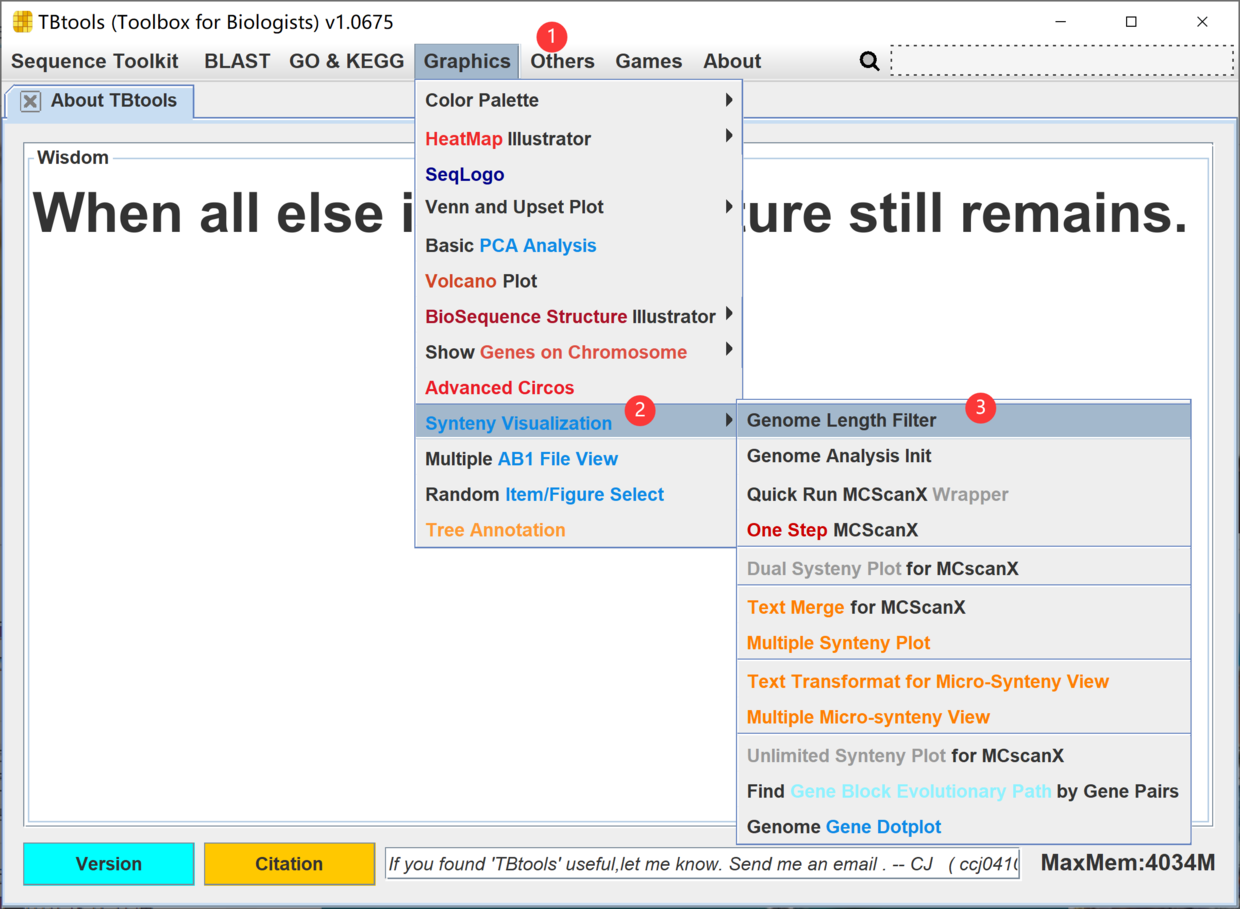
首先是获取长度信息并排序,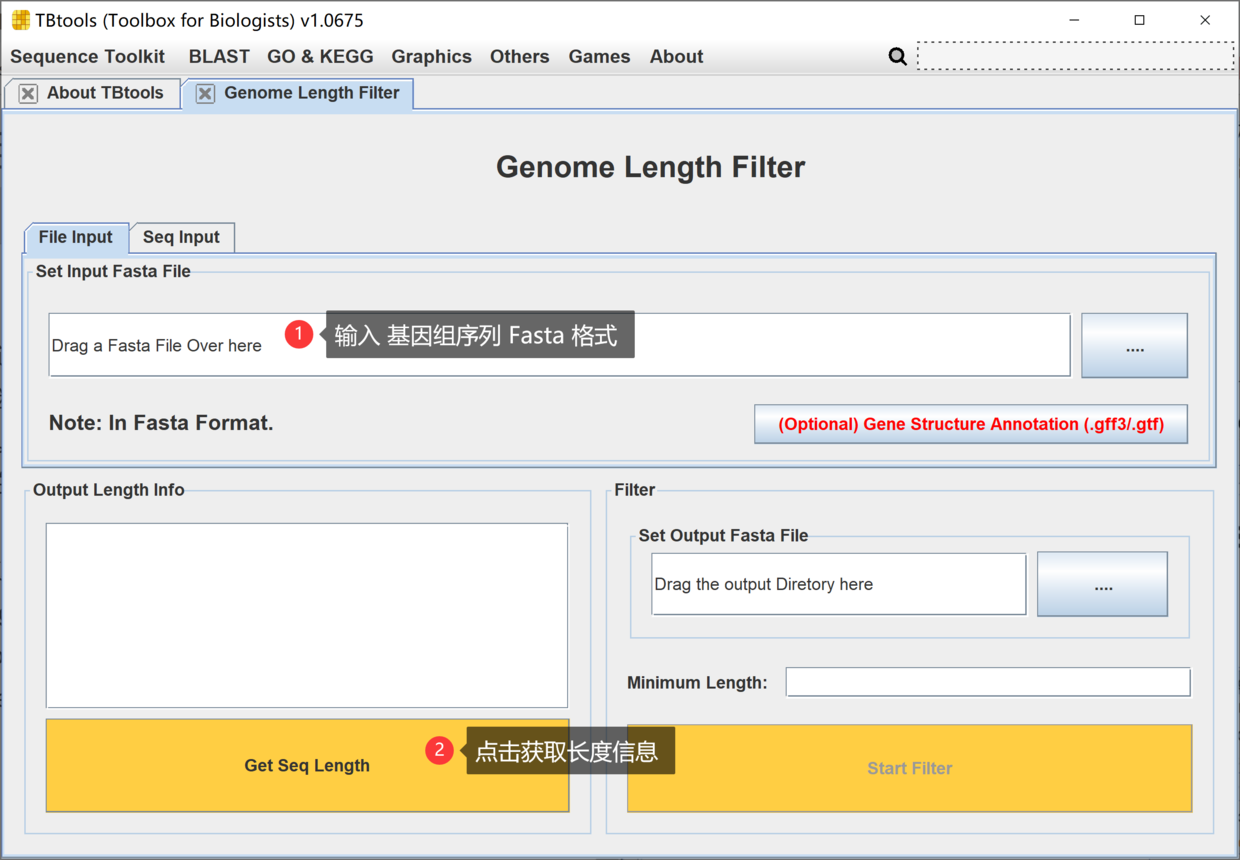
设置并运行如下,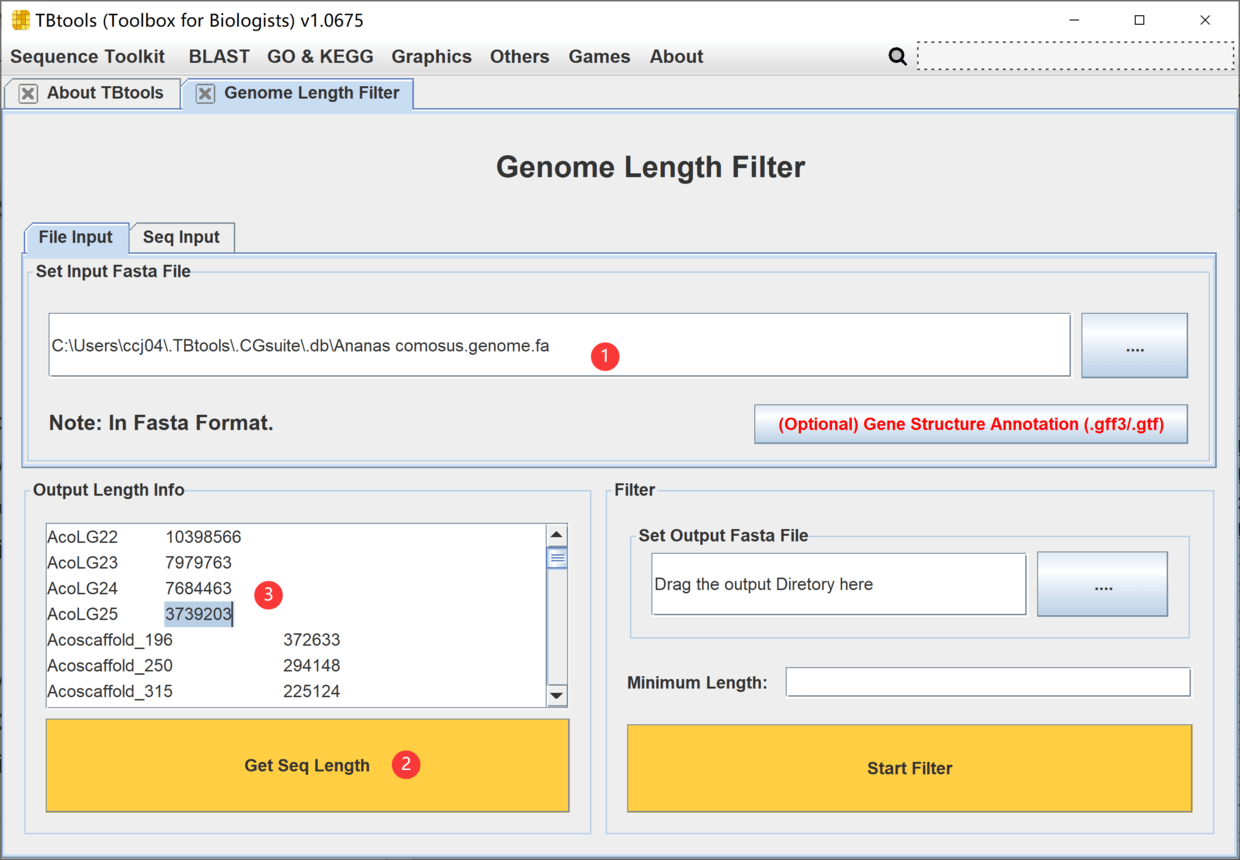
点击 Get Seq Length 之后,TBtools会获取,排序并显示所有序列长度,基于序列 ID 和长度。我们可以确定,菠萝这个基因组序列,要过滤掉碎片,那么就设置长度为 3739203。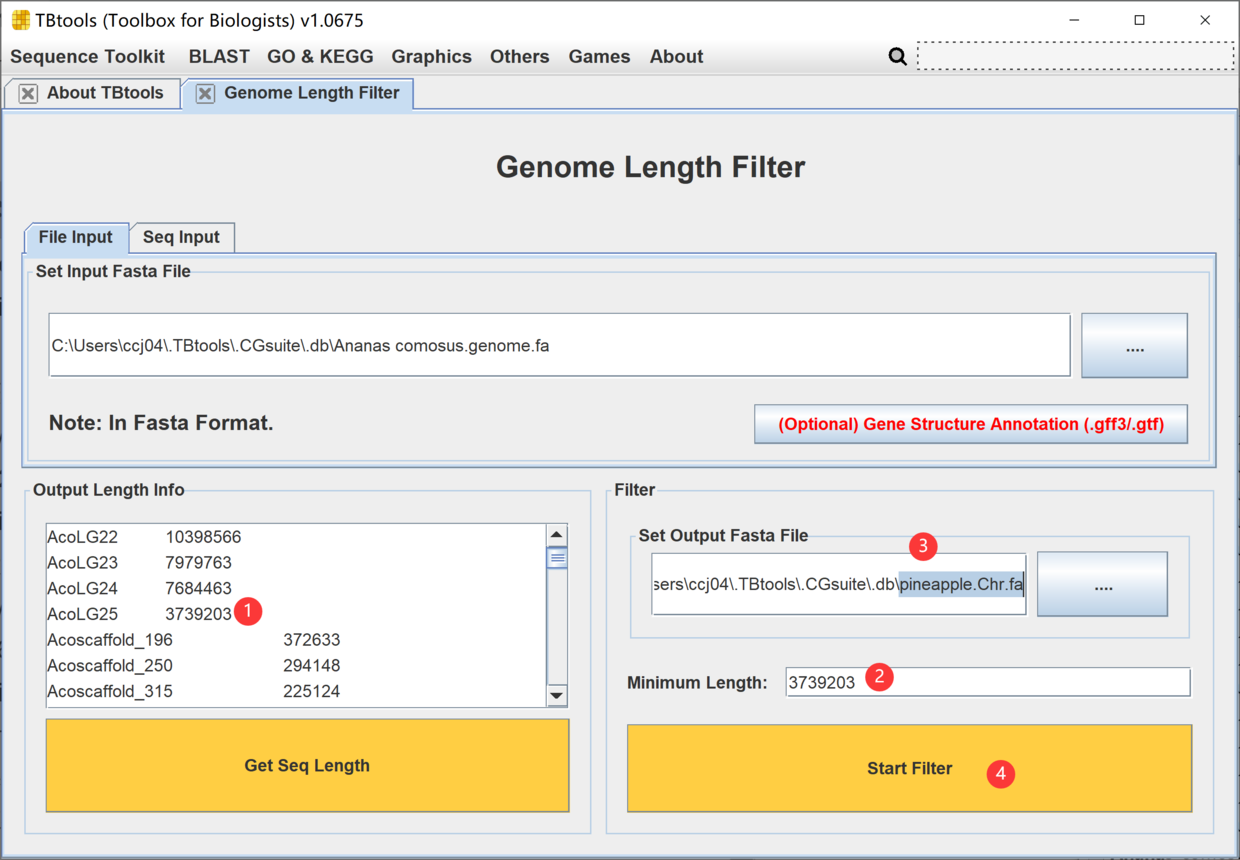
复制黏贴该长度,设置输出文件路径,随后Start Filter即可完成过滤。一气呵成。
当然,或许我们要同时过滤基因结构注释信息,那么就点击 (Optional) Gene Structure Annotation Filter,通过文件选择框,设置基因结构注释信息文件即可。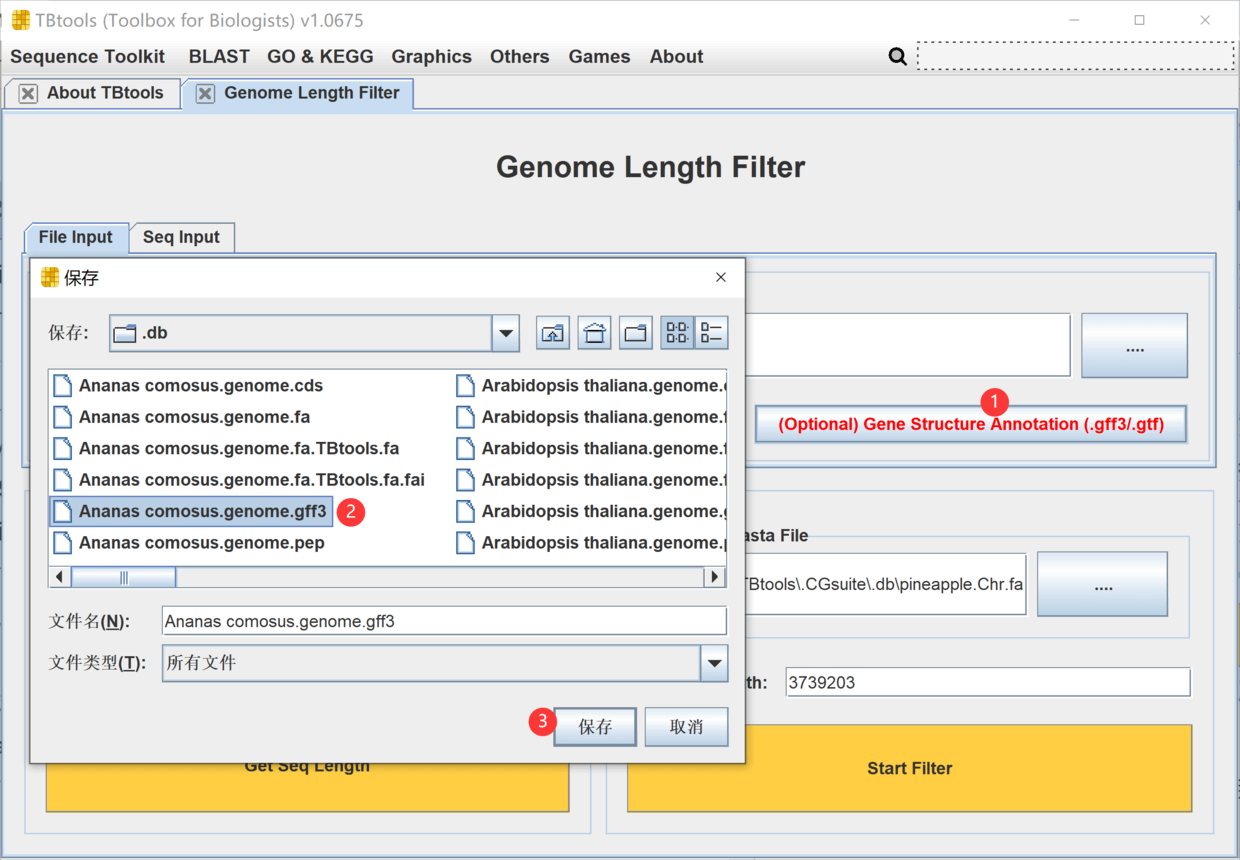
点击开始过滤,即可一次获得过滤过的 基因组序列信息 和 基因结构注释信息。
pineapple.Chr.fa # 基因组序列文件pineapple.Chr.fa.gxf # 如果输入的是gtf文件,那么输出就是gtf格式,如果输入是gff3格式,那么输出也是gff3格式...
写在最后
非常明显,这个功能其实是整合了 TBtools 已有的三个功能。尽管只是整合,我个人觉得还是非常实用。
有时候,我们并不是不知道问题的答案,而是不知道我们其实还没搞清楚问题是什么。

若有收获,就点个赞吧
0 人点赞
