基于输入的 Fasta 序列文件 ID,提取代表性序列集合,整体功能说明可以看功能界面。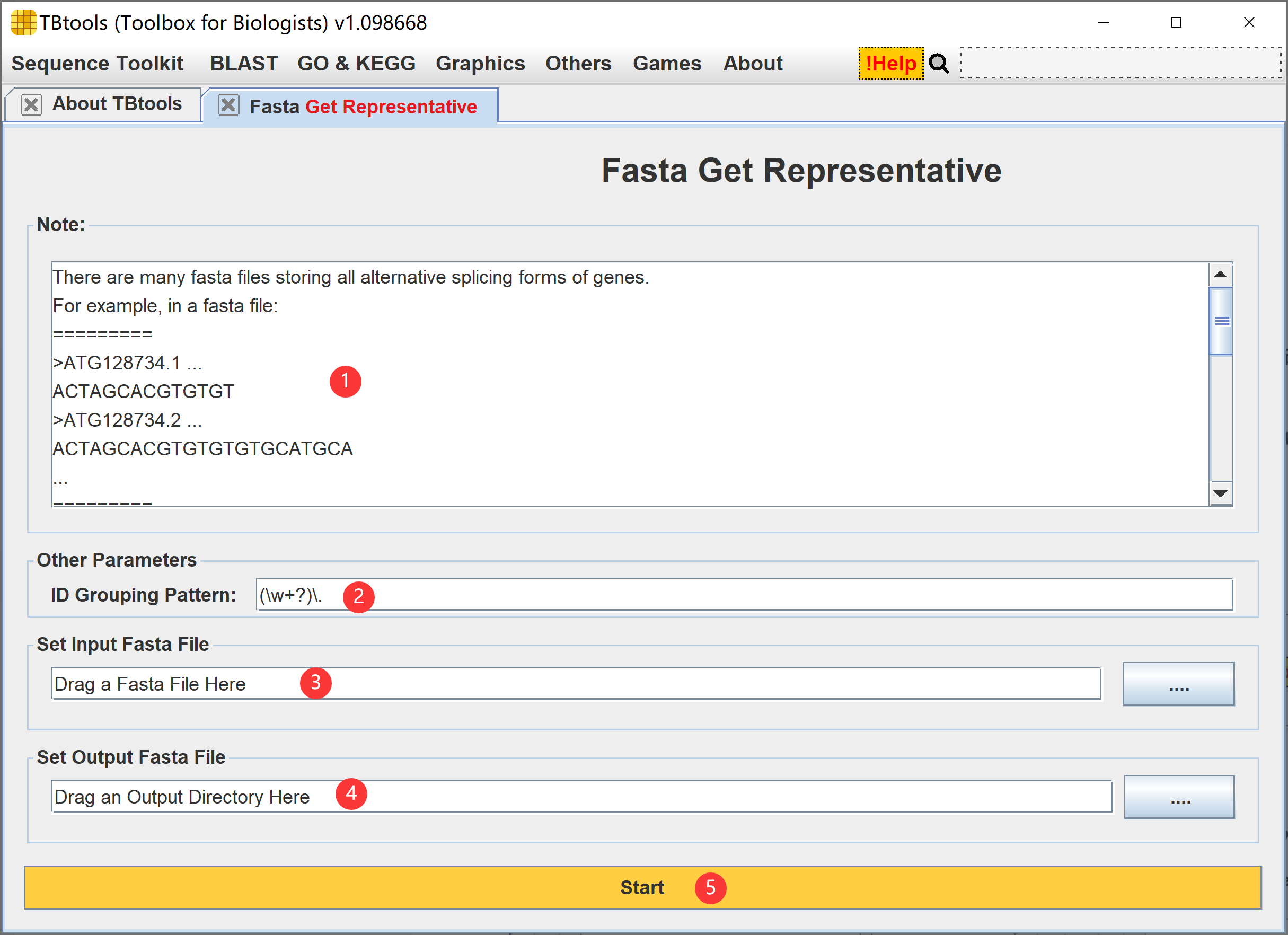
界面详解如下:
- 功能说明
- 序列 ID 归组模式
- 设置输入的 Fasta 序列文件
- 设置输出文件路径
这个功能目前只建议在无法获得物种的 GFF3、GTF 文件和基因组序列信息的情况下使用。
如输入的序列文件
ATG128734.1 … ATGGACTAGCACGTGTGT ATG128734.2 … TGGACTAGCACGTGTGTGTGCATGCA …
可以判断 .1 或 .2 等标记了同一个基因的不同转录本,可以使用归组模式「(\w+?)\」
对于其他类型,如
Trans10111 gene=Gene_1 ACGATCGACTAGCATGCATCGAT Trans10112 gene=Gene_1 ACGATCGACTAGCAT …
则需要使用新的模式,如「gene=(\w+)」来正确归组

若有收获,就点个赞吧
0 人点赞
