TBtools 中有数个序列提取功能,其中最为推荐的即这一功能。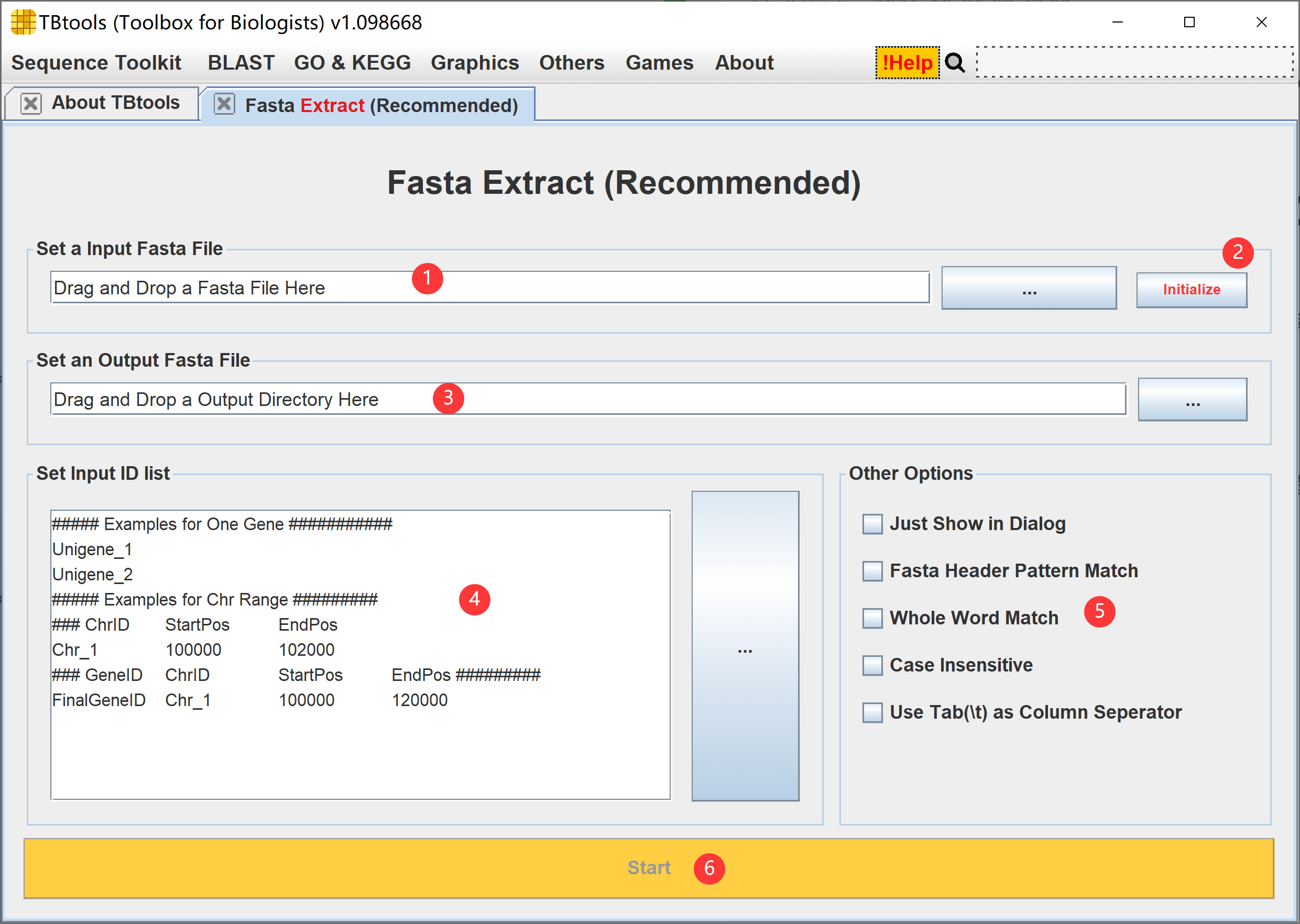
从界面上来看,分为 6 大块,其中绝大多数是可选项(即可以不做调整)。
- Fasta 序列文件输入文本框,用户可以直接拖拽硬盘中的 Fasta 文件并放置到文本框中,路径会自动获取;也可以点击跟随文本框的摁钮“…”,在弹出文件选择框中选取对应文件即可
- Initialize 摁钮,在设置 Fasta 序列文件后,可以看到 Start 摁钮仍然不可点击。需要用户点击 Initialize 摁钮,创建 Fasta 序列索引文件(如前期已有,则会软件会自动复用,节省计算时间)
- 输出文件设置文本框,用户同样可以拖拽放置文件或者文件夹,程序会自动获取输出文件夹,用户需要补全一个输出文件名;当然也可以直接点击跟随文本框的摁钮,在弹出的问价选择框中设置对应输出文件即可
- 待提取序列信息设置框,参考界面说明,接受三种类型的提取模式:
- 基于 ID 的完整序列记录提取,如输入 Unigene_1 … 等完整序列 ID,每行一个,即可提取完整序列记录
基于 序列坐标信息,进行序列区间截取,如提取染色体 Chr1 上第 10000 个碱基到 20000 个碱基的一段序列,那么输入如下。如果需要提取反向互补序列,使起始坐标大于终止坐标即可。
# 注意,制表符[\t]分隔,而非空白[Space]分隔Chr1 10000 20000# 提取反向互补序列,则翻转碱基坐标Chr1 20000 10000
提取坐标信息的功能,重命名区间,如我们需要提取 Peak 或者 Promoter 序列信息,并指定输出时序列名字
peak_1 Chr1 10000 10200promoter_ATG8 Chr2 20300 22300
- 系列可选参数

- Just Show in Dialog,即提取结果不输出到文件,直接弹出文本对话框,显示提取结果,这一选项对于少量序列提取有较大便利
- Fasta Header Pattern Match,对于一些 Fasta 序列文件,其 ID 不能可能会有注释信息,如“>Unigene_1 MYB101 protein,transcription factor”。我们可以使用“MYB”作为输入 ID,从而提取所有 MYB 序列
- Whole Word Match,主要解决输入“MYB10”可同时匹配“MYB10”、“MYB101”、“MYB102”等情况,勾选这一选项,即可只匹配“MYB10”
- Case Insensitive,很多时候,注释文件甚至是 ID 会存在大小不同,尤其是“myb”需要匹配“MYB”和“myb”时,那么勾选这一参数
- Use Tab(\t) as Column Seperator,默认情况下,为了支持部分用户的输入习惯,TBtools 同时支持制表符和空白符,但有时候,用户或许希望重命名输出序列的 ID 含空白符,如“Myb 101 Promoter Chr10 100000 102000”,那么需要勾选这一参数。
- 一切准备就绪,点击 Start 即可。
这一功能唯一小缺点是 会自动建立Fasta文件索引,需要占用一点硬盘空间。

若有收获,就点个赞吧
0 人点赞
