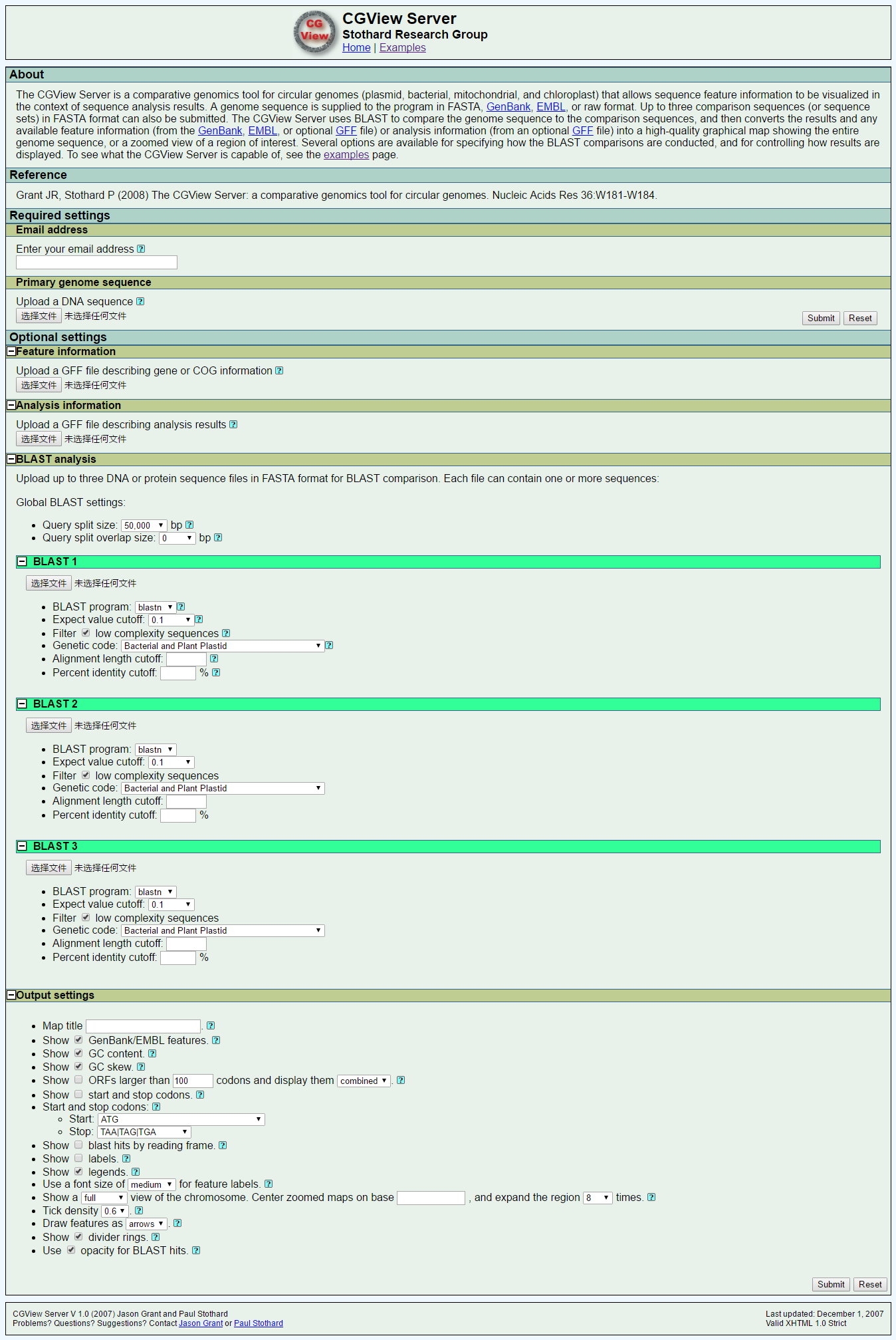
1. 软件网络上的截图(点击图片放大更清晰)
(软件地址:http://stothard.afns.ualberta.ca/cgview_server/)
2. 使用简介
(1)前期介绍
①CGView可以使任何细菌,质粒,染色体,或者线粒体的基因特点可视化。可以帮助鉴别保守的基因片段,基因的水平转移,基因上拷贝数量上的差别。
②通过相似性比较基因。
③PipMzker和ACT可以去可视化用户提供的序列相似度。PipMzker是一个网站服务器计算同一性百分率,可以显示百分率和空位(gap)。
④ATC是独立于java的一个程序,和blast联合使用比较基因序列。先要将序列blast, ATC再将Blast后的序列用有颜色的区域将相似的序列标记。这些线条可以显示哪段基因是保守的,可以将基因的不同结构区域标亮。
⑤尽管ACT和PipMaker可以处理来自任何数据库的序列,但是在可视化细菌和细胞器序列上不受欢迎,其他的程序像CGView, GenomePlot, GenoMap 和 Microbial Genome Viewer就可以用了。
(2)程序说明书及各参数介绍。
①网站上提交要分析的数据,至少要提供DNA序列和邮箱地址。
可以提交4种格式的序列:raw, FASTA,GenBank 和 EMBL格式。如果使用GenBank 或 EMBL,文档里的基因注释将会出现在图中。邮箱地址是需要的,因为分析需要一段时间,分析完后发送到你的邮箱。
②增加一个附属的处理原始DNA序列的功能—GFF文件,GFF是用来描述与核苷酸及蛋白质序列相关的基因和其他特点。这种特点可以在所做图中基因的相应位置显示出来。GFF在“feature”栏包含一个COG分类,CGView会根据COG分类把特点标颜色。除了使用COG分类上色外,还可以根据其他基因类型比如CDS,tRNA,rRNA等等进行分类上色。
③GFF文件可以来源于多种分析程序,比如Excel.
④数量统计也可以放在图中,通过再次使用GFF文件。这个函数可以可视化其他分析程序的得分或者测量结果。
⑤为了增加的DNA序列比对的需要。最大可以使用3个用来blast的数据库,可以使用fasta或者multi-FASTA,多FASTA格式允许使用序列集合进行单一比较。默认的数据库包括所有蛋白质家族的成员,或由特定细菌基因组编码蛋白质组。
⑥对于每一个比较组(blast1,blast2,blast3)里有一组选项指定搜索类型和搜索参数。允许在DNA和蛋白质水平上进行搜索。并根据重要性(e值)进行过滤,对齐长度和百分比同一性。
⑦CGView Server界面的最后一部分是设定序列分析后的特点展示。
(i) 如GC content,GC skew, ORFs, 起始和终止密码子。去调整图像上的结构。
(ii)比如“blast hits by reading frame”可以根据阅读框计算,这种能力可用于鉴定哪些ORF在重叠组中是保守的。
(iii)“opacity for BLAST hits”还可以用部分不透明度绘制,使一级序列中多个重叠被识别。
(iv)其他选项包括绘制缩放的图形,特征标签、图标,和标题。
(3)程序计算原理。
提交给CGView服务器的数据进入分析
队列。 Perl程序定期检查队列,开始处理。处理开始于使用的formatdb程序(包含在BLAST中),将任何比较序列转换为BLAST数据库。
作为查询的主要序列,在调用独立BLAST之前。首先将其拆分为用户定义的较小子序列。序列文件,BLAST结果,GFF文件和用户选项传递给另一个构建XML的Perl脚本,CGView地图绘制程序的文件。 CGView生成PNG图像,图像和描述提交的文件和设置通过电子邮件发送给用户。
(4)图形结果解释。
①圆环用于展示从传入序列里读取到的基因信息。特点和分析结果来自于GFF文件,ORFs, 起始密码子和blast结果构成基本图。
②特征根据类型着色,并且在某些情况下,调整特征的高度以反映它的属性。在“blast hit”中,高度与同一性大小成正比。相似的,GFF文件里得分值用于确定图形里的高度。
③另外一个选项图标,去将所有的特征上颜色。
④图片中的标签根据特点绘画,来自于传入的序列或者GFF文件。
⑤一个序列标尺,画在特征环内最里面的,允许要确定的特征的近似位置。
(5)软件缺点
与ACT这样的独立工具相比,CGView Server的一个缺点是是服务器返回静态图片,虽然这些图像适合出版,ACT可能对深入探索序列和BLAST结果更有用。
(6)美图欣赏(点击放大超清)
以下图片均来自网站示例数据。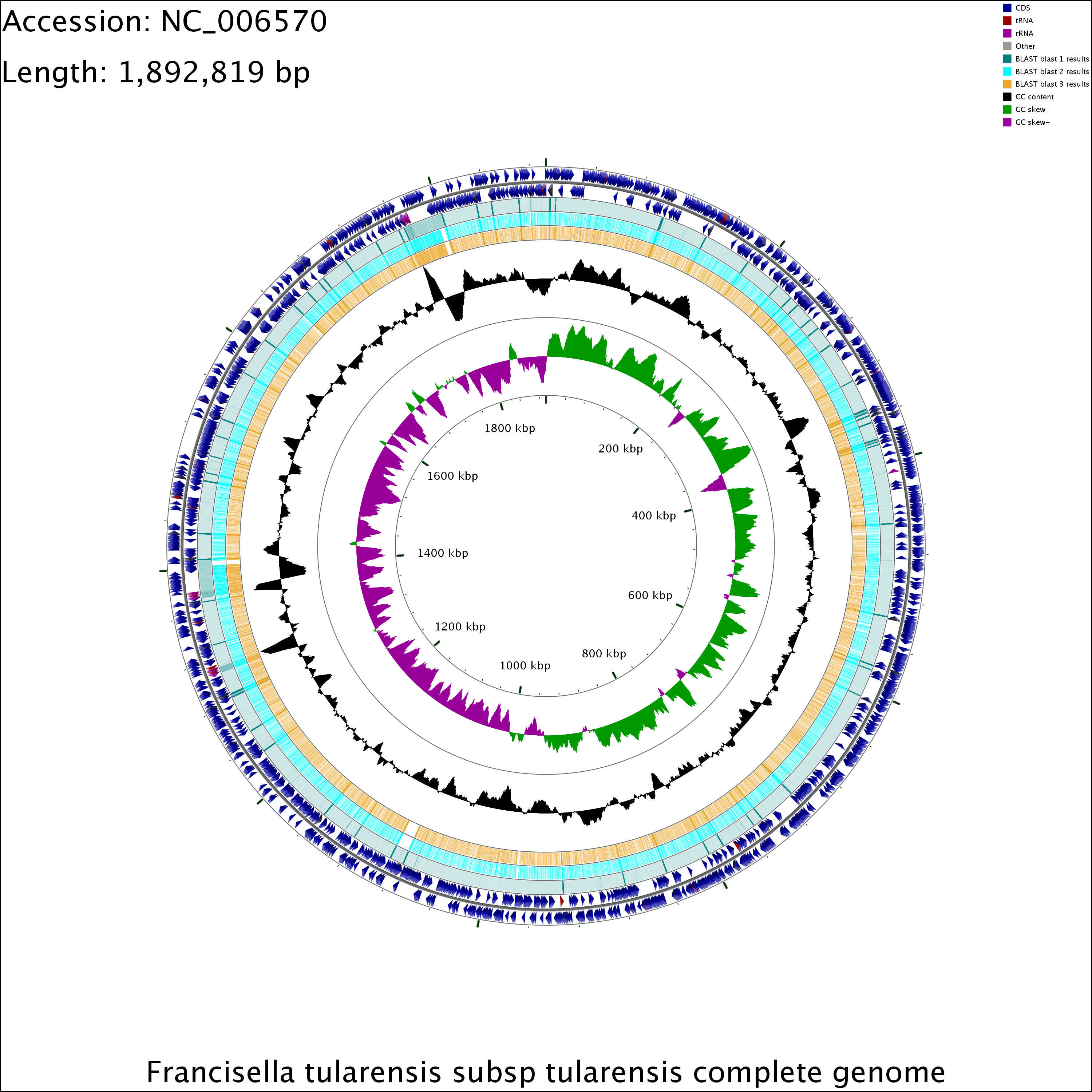
转载请注明出处。

若有收获,就点个赞吧
0 人点赞
