一、序列比对的两种策略
1.全局比对(global alignment)
它是假设两条序列在整个长度上是相似的,然后从头到尾比较两条序列的最佳匹配,适合高度相关等长序列比对。不适用于发散的不同长度的序列,因为它不能识别两条序列中高度相似的局部序列。
2.局部比对(local alignment)
不考虑两条序列全局相似,而是找两条序列中高度相似的局部区域而不考虑其他区域,适合包含相似模块分散的生物序列,可以找出domain(保守区)和motif。
3.两种策略的区别
假设如下两条序列Query和Subject,分别采用全局比对和局部比对比对策略,从下图红色方框里可以看出差异:

4.有无Gap比对
两条序列,进行有空位比较和无空位比较的差异如下图:
二、序列比对算法
不管采用哪种策略,比对算法基本上是相似的:
- 点阵方法:构造一个二维矩阵。很容易识别序列中高度相似的区域,它在识别染色体重复(两条相同序列,即自己和自己比)和比较两个高度相关的基因组中的基因顺序的保守性非常有用。缺点:很难构造多序列比对。
动态规划方法:匹配两条序列中所有可能字符,也是构造一个二维矩阵确定最优比对方法。里面引入“空位罚分”,即代表插入和删除的空位,因为在自然进化中插入和删除的发生频率比替换相对少。而且开始一个新的空位和拓展一个已经存在的空位付出的代价是不同的(原理是一旦插入和删除发生,那么临近的一些残基很容易插入和删除),两者会有不同的罚分。序列末尾不进行罚分,因为实际中很多同源序列是不等长的,如果末端进行了空位罚分反而是不实际的结果。
启发式算法
一个基于直观或经验构造的算法,在可接受的花费(指计算时间和空间)下给出待解决组合优化问题每一个实例 的 一个可行解,该可行解与最优解的偏离程度一般不能被预计。
详细见后。
三、常用的序列两两比对方法
1.全局比对的动态规划算法
经典的算法是Needleman-Wunsch算法。它必须对序列从头到尾进行计算得到最高比对得分。缺点,关注全长的最大比对的缺点是找不到局部的序列相似。对于发散序列和具有不同结构域的序列,这种方法达不到最理想的比对。全局两两比对的web程序是GAP.
2.局部比对的动态规划算法
正常比对序列中,两条被比对序列的分离水平是不知道的,两条序列的长度也可能不同,这种情况下,识别局部相似性比去对比包含残基的整个序列更加有意义。第一个运用动态规划进行局部序列比对的算法是Smith-Waterma算法。<br /> 这种算法下的局部比对分为两种:<br /> local alignment (Smith-Waterman) with affine gap costs (Gotoh)、local alignment with generalized affine gap costs (Altschul)<br /> 全局比对中,最终结果受到选用得分矩阵的影响,而局部比对的目标是找到局部最高分。这种方法适应于对分散序列和具有来自多个不同源的区域序列,目前大多数两两比对程序基本采用局部比对策略。
四、多序列比对算法
1.动态规划算法
可得到最优解,但是计算量非常大,实际中很难用于多序列比对。
2.启发式算法(heuristic algorithm)
(1)渐进法(progressive methods):Clustal, T-Coffee, MUSCLE
(2)迭代法(iterative methods):PRRP, DIALIGN
(3)其它算法:Partial Order Algorithm、profile HMM、meta-methods (MAFFT)…
注:启发式算法的计算过程也有可能利用到动态规划算法,比如Clustal采用的渐进式算法: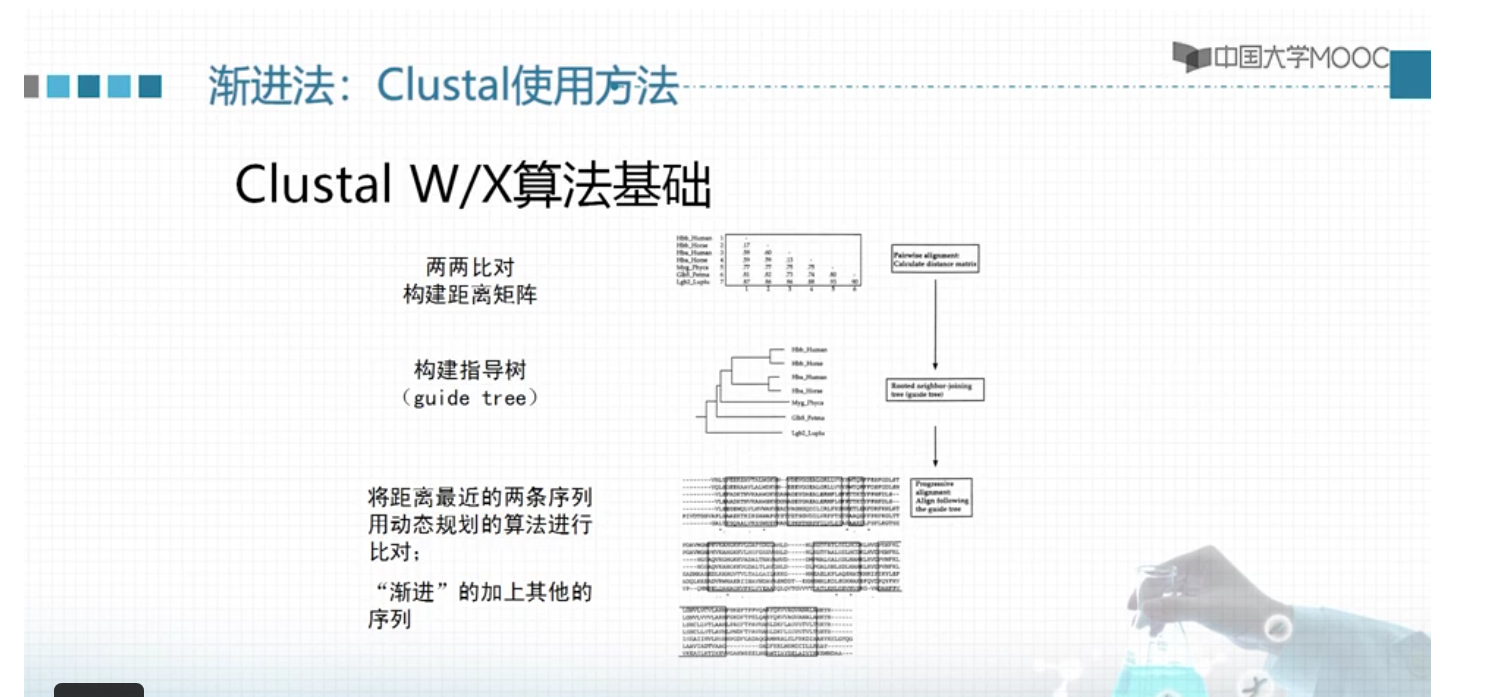
五、比对中用到的打分矩阵
1.核苷酸得分矩阵
核苷酸得分矩阵相对简单,对匹配位置赋予一个正值或者高分,对失配位置赋予一个负值或者低分。但是这种方法不符合实际,观察显示转换(嘌呤与嘌呤或者嘧啶与嘧啶之间的替换)发生频率比颠换(嘧啶与嘌呤之间的替换)高,因此,需要一个反应不同残基替换发生频率不同的更加复杂的统计模型。
2.氨基酸的替换矩阵
氨基酸的替换矩阵比较复杂,某种氨基酸很容易被具有相似理化性质的其他氨基酸替换,而很难被具有不同理化性质的氨基酸替换。而不同理化性质的氨基酸替换可能导致结构和功能的缺失,这种会导致分类的替换是很少被进化所选择的。<br /> 氨基酸替换矩阵是一个20*20矩阵,它用来反映氨基酸被替换的可能性。 经验上的矩阵,包括PAM矩阵和BLOSUM矩阵。
(1)PAM矩阵
PAM的意思是可接受的点突变,观察到的突变不会改变蛋白质的一般功能,观察到的氨基酸突变被认为是自然选择接受了的。
一个PAM单位被定义为有1%的氨基酸位点发生了变化或者100氨基酸有1个发生突变,但这并不意味100次PAM后,每个氨基酸都发生变化,因为其中一些位置可能会经过多次突变,甚至可能会变回到原来的氨基酸。一个特定残基对的PAM分数是通过一个多阶段过程得到的,这个过程包括:
①计算相对突变率。一个特定氨基酸被同源氨基酸替换的总数除以在整个比对中这种氨基酸出现的总数;
②用随机替换率对预期的氨基酸替换率进行标准化,把标准化的突变率除以特定氨基酸出现频率,然后取以10为底的对数。把结果取整后填入替换矩阵,这个矩阵就可以反映氨基酸替换的可能性。
③对于较分散序列的高阶PAM矩阵是通过对PAM1矩阵相乘推出来的,例如,PAM80就是对PAM1矩阵自乘80次得到的。一个PAM80矩阵只相当于观察到突变率的50%,PAN250表示一致性为20%。
缺点:PAM矩阵构建过程中,只是之间观察了PAM1中的基于一小堆极近相近序列中的残基替换(这个要从PAM矩阵的来源说起,当时构建这个矩阵用的序列是21组非常接近的蛋白质序列),PAM矩阵对于分散序列的比对是不可靠的。
PAM矩阵数字与序列相似度的对应关系:
| 序列相似度 | 打分矩阵 |
|---|---|
| 20% | PAM250 |
| 40% | PAM120 |
| 50% | PAM80 |
| 60% | PAM60 |
注意:PAM250矩阵 → 对应这些用于比对的序列相似度估计在14% – 27%,一般PAM250矩阵对应序列相似度在20%。
(2)BLOSUM矩阵
为了弥补PAM的缺点,BLOSUM矩阵应用而生,BLOSUM矩阵与PAM矩阵的不同之处在于:用于产生矩阵的蛋白质家族及多肽链数目,BLOSUM比PAM大约多20倍。BLOSUM矩阵不用推断,而是用实际上所选序列的残基一致性的比例来构建矩阵。
与PAM矩阵的阶数相反,BLOSUM矩阵阶数越低代表序列越分散。
比如BLOSUM60,这个矩阵首先寻找氨基酸模式,即有意义的一段氨基酸片断(如一个结构域及其相邻的两小段氨基酸序列),分别比较相同的氨基酸模式之间氨基酸的保守性(某种氨基酸对另一种氨基酸的取代数据),然后,以所有 60%保守性的氨基酸模式之间的比较数据为根据,产生BLOSUM60;同理,以所有80%保守性的氨基酸模式之间的比 较数据为根据,产生BLOSUM80。
(3)两个矩阵的比较
①PAM1矩阵是通过一个进化模型得到的,而BLOSUM矩阵完全是由观测值构成,因此,BLOSUM矩阵可能没有PAM矩阵那么强的进化上的实际意义,这也是PAM矩阵被常用来重构系统发育树的原因。
②PAM矩阵对发散序列可能不符合实际。
③BLOSUM矩阵是完全通过保守序列的局部比对得到的,而PAM1是对包含保守和变化区域的整个序列全局比对得到的。这是BLOSUM矩阵更加适用于搜索数据库和寻找蛋白质中保守区域的原因。一些经验上测试BLOSUM矩阵在局部比对的正确性上胜过PAM矩阵。
对于一个给定的矩阵,一个正的分数说明在一个同源序列的数据集里观察到的氨基酸替换频率比随机替换频率高;零分表示在一个同源序列的数据集里观察到的氨基酸替换频率和随机替换频率相等;负数一个同源序列的数据集里观察到的氨基酸替换频率比随机替换频率低,这通常发生在不相似残基之间。
六、序列比对的统计学显著性
序列比对本身就是一个随机问题,我们要对这种随机进行检验,比对完后,会有一个P值。
P值的解读:
- 如果P值小于10,表明两条序列是精确匹配的;
- 如果10<P<10,表明两条序列近似匹配;
- 如果10<P<10,表明两条序列有较近的同源关系;
如果10<P<10,表明两条序列可能存在较远的同源关系;
如果P>10,那么这两条序列能匹配上可能是由于随机的关系;
可以使用软件PRSS,用来评价序列两两比对在在统计学上显著性。
七、序列的同源性和同一性(Identity)
同一性和同源性这两个词经常被混淆。同源性只有高低之分,没有具体数值,属于”质”的属性;而同一性才有具体数值,是“量”词。同一性数值越大,同源性越高。

若有收获,就点个赞吧
0 人点赞
