该篇为介绍MultiTypeTree进行结构溯祖的教程
转载来源:https://github.com/tgvaughan/MultiTypeTree/wiki/Beginner’s-Tutorial
额外附件:Efficient Bayesian inference under the structured coalescent.pdf
Beginner’s Tutorial
Tim Vaughan edited this page Jul 27, 2016 · 48 revisions
Introduction
In this tutorial, we will use the BEAST 2 package MultiTypeTree to perform a Bayesian phylogenetic analysis of an influenza data set using the structured coalescent model.
The data set used in this tutorial is a thinned (150 sequence) subset of the (980 sequence) data set used in the publication Vaughan et al., 2014, which in turn was assembled from publicly-available data sets provided by various authors on GenBank.
Software Requirements
In order to proceed, ensure you have the latest version of BEAST 2 installed. To analyze the inference results you’ll also need a recent versions of Tracer and FigTree.
Installing the MultiTypeTree package
You can easily install this package via BEAUti’s package manager. To do this, follow these steps:
- Start BEAUti
- From the File menu, select Manage packages.
- Find “MultiTypeTree” in the list of packages shown, select it and then click “Install/Upgrade”:
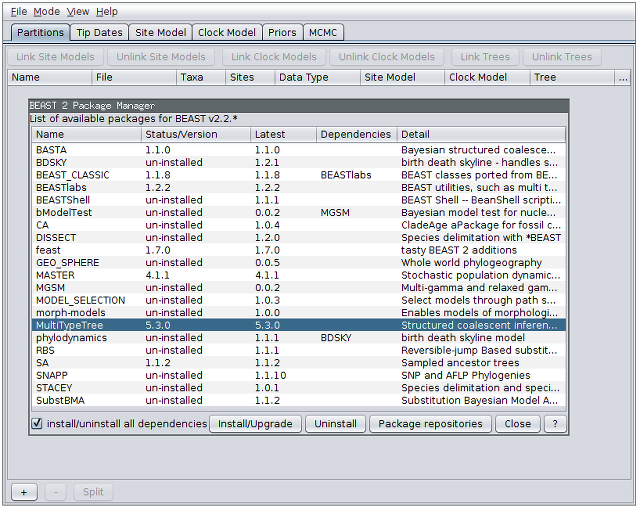
Finally, restart BEAUti. This is very important. Strange behaviour will result if you do not restart the program.
Setting up the analysis using BEAUti
Loading the Template
From the File menu, select Template and then choose MultiTypeTree: 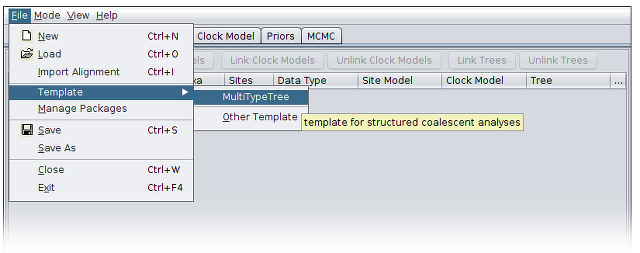
Loading the data
Once the template is loaded, we can load in our example sequence data. In our case, this data is stored a FASTA file, the first few lines of which look like this:
> CY003176USA_2002.11780822
—————-GGGATAATTCTATTAACCATGAAGACTATCATTGCTTTGAGCTACATTT
> CY000441_USA_2002.0109589
—CAAAAGCAGGGGATAATTCTATTAACCATGAAGACTATCATTGCTTTGAGCTACATTT…
> CY019141_USA_2004.97808219
—————————TTCTATTAACCATGAAGACTATCATTGCTTTGAGCTACATTC…
> CY001096_USA_2003.9260274
——————————-TATTAACCATGAAGACTATCATTGCTTTGAGCTACATTC…
> CY010084_NewZealand_2005.62739726
——————————-TATTAACCATGAAGACTATCATTGCTTTGAGCTACATTC…
> CY007387_NewZealand_2004.63287671
——————————-TATTAACCATGAAGACTATCATTGCTTTGAGCTACATTC…
> CY012432_NewZealand_2000.81643836
—————————————-CCATGAAGACTATCATTGCTTTGAGCTACATTT…
The lines beginning with “>” are labels for the sequences immediately following. In general, these labels have no special format, but in this file each label is a underscore-delimited triple. The first element of each triple is the GenBank accession number of the sequence, the second is the geographical region from which it was sampled, and the third is the time at which it was sampled measured in calendar years or fractions thereof. (The ellipses are not in the file, but are used here to indicate that sequence has been truncated.)
In this tutorial we will be using the influenza sequence data which is distributed with MultiTypeTree. To make this easy to find, open the _File menu and select Set working dir. Then, from the submenu that appears, select MultiTypeTree. 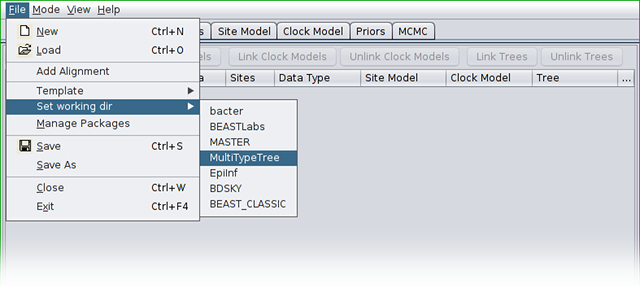
(This step is not required when loading your own data.)
To load the file, open the File menu and select Add alignment. This will open a file selection dialog box. The example influenza sequence data file is named h3n2.fna. Assuming you have followed the previous step to set the working directory, this can be found in the examples/ directory shown when the file selection dialog box loads.
Once this file is loaded, your BEAUti screen should look something like the following: 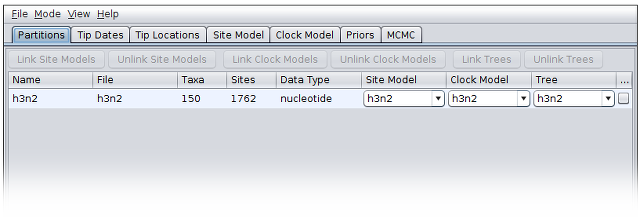
Setting up dates
Once the data is loaded, the next step is to specify the times at which the sequences were sampled:
- Select the “Tip Dates” panel.
- Check the “use tip dates” checkbox.
- Click the “Guess” button at the top-right of the panel. This opens a dialog that allows sample times to be loaded from a file or inferred (guessed) from the sequence labels.
- Because the times are included as the last element of the underscore-delimited triple in the header, choose the “use everything” radio button and select “after last” from the drop-down menu. (Note that the underscore character is already chosen as the delimiter.)
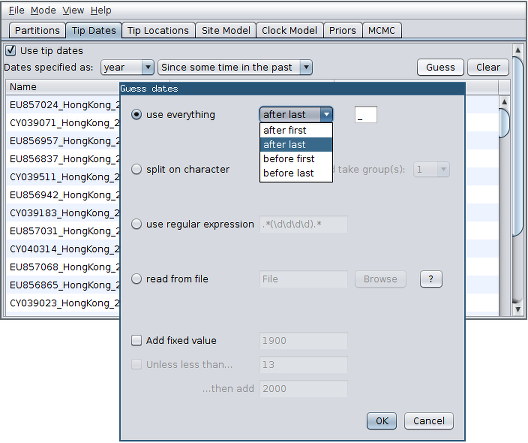
After clicking “OK” you should find that the tip date table is populated with times that match those in the sequence headers, and that the last column of the table contains “heights” (times before most recent sample) calculated from the times: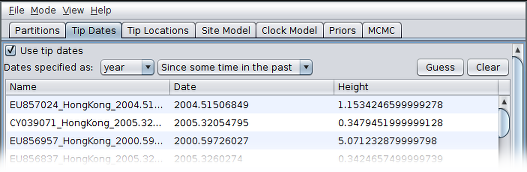
Setting up locations
Now we’ve specified the sampling times, we move on to specifying the sampling locations. To do this, we follow a very similar set of steps to those we used to set the sample times:
- Select the “Tip Locations” panel. You’ll find that the locations are already populated with default values.
- Click the “Guess” button at the top-right of the panel. This opens the same dialog that we saw in the previous section.
- Because the locations are included as the second element of the underscore-delimited triple in the header, choose the “split on character” radio button and select group 2 from the drop-down menu. (Note again that the underscore character is already chosen as the delimiter.)
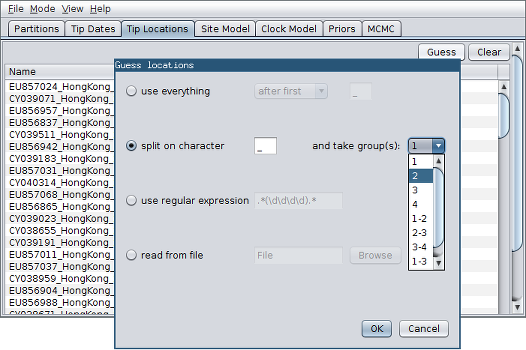
After clicking “OK” you should find that the tip location table is populated with locations that match those in the sequence headers, as follows: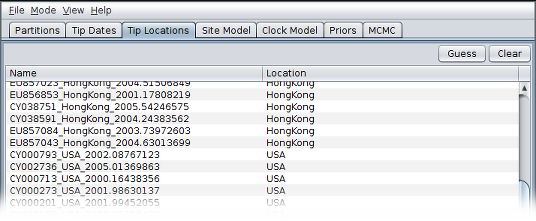
Substitution model
For this analysis, we use a GTR substitution model with an unknown (estimated) proportion of invariant sites. To configure this in BEAUti:
- Switch to the “Site Model” panel.
- Select GTR from the drop-down menu (the default option is JC69).
- For the “Proportion Invariant” enter “0.5” (or really any other number greater than 0 and less than 1) and check the adjacent “estimate” checkbox.
The BEAUti panel should now look like the following: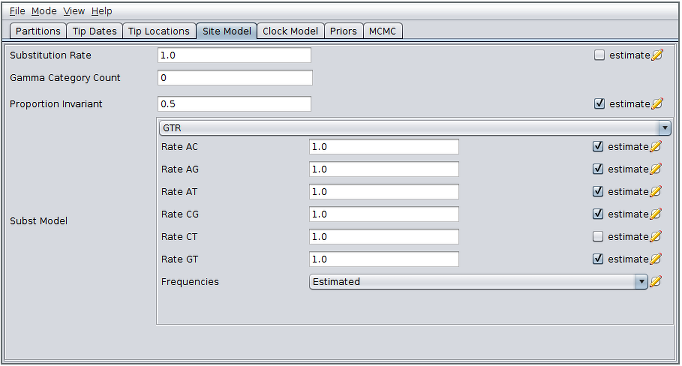
Note that the unchecked estimate box next to the CT rate is important to maintain identifiability. (All the other transition rates can be considered relative to the CT rate.) Note also that the “Substitution rate” defined on this panel should be left unestimated for a similar reason - we use the “Clock rate” defined in the “Clock Model” panel to determine the average per unit time rate of sequence evolution. Used this way, the “substitution rate” is therefore not actually a rate (it’s actually dimensionless) but is instead a rate multiplier that in our case we fix at 1.
Defining Clock model
For this analysis we assume a strict clock. The default values given in the “Clock Model” panel are therefore sufficient. (By default the Clock.rate parameter is estimated whenever sequences are serially sampled.)
Adjusting Priors
Because the structured coalescent is a model-based prior on the (multi-type) tree distribution, setting up the “Priors” panel is a particularly important part of setting up this analysis.
- Click the arrow to the left of the structuredCoalescent.t:h3n2 parameter to expand the tree prior.
- Click the up-arrow on the “number of demes” spinner to change its value to 3, matching the number of discrete locations in our model.
- Check the “estimate” checkboxes to the right of the population size vector and migration rate matrix tables. (These tables allow us to modify the initial values of these parameters, or fix them to known values.)
- Use the drop-down menus to change the priors for the clockRate.c:h3n2, popSizes.t:h3n2and rateMatrix.t:h3n2 parameters to “Log Normal”.
- Click the arrows on the left-hand side of each of these parameters to alter the details of these priors, changing the M parameter to 0 and the S parameter to 4.
With the parameter priors de-expanded, the panel should finally appear as below: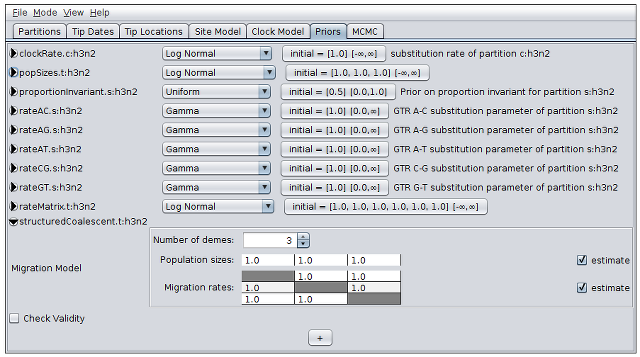
MCMC Settings
In the MCMC panel, we can adjust the number of steps to use in the MCMC chain that will be used to sample from the joint posterior over multi-type trees and all of the model parameters. In this case, we will stick with the default of 10 million steps.
This panel also lets us adjust the fraction of sampled states that are written to the log and tree log files.
Notice that the MultiTypeTree template provides two special log types: the first is “maptreelog”. This produces a file containing running estimates of the maximum a posteriori multi-type tree over the course of the analysis. For this particular log type, the sampling period defined in the “Log Every” field also determines the fraction of trees that will be assessed as MAP tree candidates. The default value therefore causes only 1 tree in every 10000 to be considered for this position: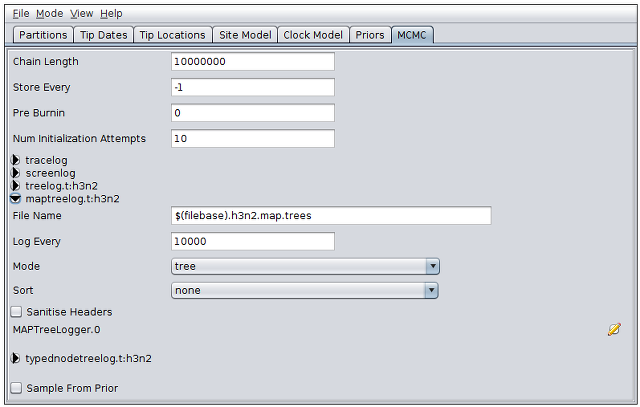
Using smaller sampling periods will improve the accuracy of the MAP estimate at the expense of increased time spent writing trees to disk and increased log file size.
The second MultiTypeTree specific log type is the “typednodetreelog”. This records trees in which only the coalescent nodes are annotated with ancestral locations. The log file can be analyzed using TreeAnnotator to produce a summary tree.
Saving the analysis specification
At this point we have fully specified our analysis in BEAUti. To actually run it, we need to save the model to an XML file and then run BEAST on that file. To save the model, simply select Save from the File menu:
Then enter a useful filename. For this analysis, something like “h3n2” will suffice. BEAUti will automatically append “.xml” to the file name you specify.
Running the analysis using BEAST
To run the analysis, simply start BEAST 2 in the manner appropriate for your platform, then select the file you generated in the last section as the input file. (Refer to the documentation provided atwww.beast2.org for detailed instructions.)
The 10 million steps require a bit over an hour to complete on a modern desktop computer. However, once the analysis is running you can immediately begin looking at the results by following the instructions in the next section.
Analyzing the results
The results of the analysis primarily consist of two parts:
- The parameter log, which is written to the file h3n2.log.
- The tree log, which is written to h3n2.h3n2.trees.
In addition, the file h3n2.h3n2.map.trees contains the running estimate of the MAP tree as a function of MCMC step number, while the file h3n2.h3n2.typedNode.trees is the TreeAnnotator-compatible file we’ll use to assemble a summary tree.
Parameter log file analysis
We can use the program Tracer to view the parameter log file. To do this, start Tracer and then press the “+” button in the top-left hand corner of the window (under “Trace files”). Select the log file for this analysis (h3n2.log) from the file selection dialog box. The “Traces” table will then be populated with parameters and summary statistics corresponding to our structured coalescent analysis.
Important traces are:
- migModel.t:h3n2popSize*: These give the (effective) structured coalescent population sizes for each of the locations in your model.
- migModel.t:h3n2_rateMatrix_A_B: These give the structured coalescent immigration rates from B to A.
- Tree.t:h3n2_count_A_to_B: these give the actual number of ancestral migrations from location A to location B (backward in time).
The panels tabs at the top-right of the window can be used to display one or more selected traces in various ways. For example, selecting the three population size traces and choosing the “Marginal prob distribution” panel results in the following useful comparison between the sampled population size marginal posterior distributions: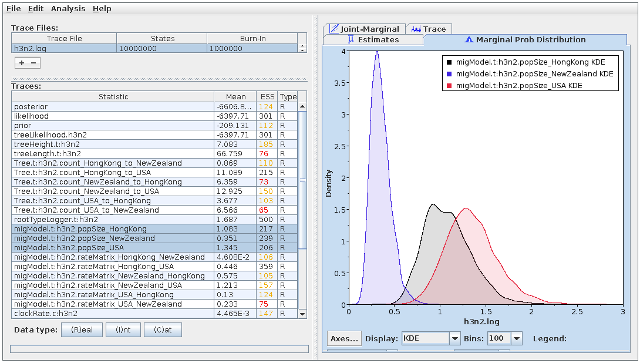
Observe that the effective sample size (ESS) estimates in the above figure are still very low, even after all 10 million steps have been taken. This is especially true for the migration rate matrix elements, and means that these results should not be trusted until many more samples have been collected. (In the published analysis, nearly 400 million steps and more than 6 times as many sequences were used.)
Tree log visualization
The popular phylogenetic tree visualizer FigTree can be used to visualize the sampled trees contained in the tree log h3n2.h3n2.trees and the MAP tree estimate log h3n2.h3n2.map.trees. Be warned, however, that FigTree currently takes an extremely long time to load even relatively small (a few megabyte) MultiTypeTree logs.
For this reasons we suggest using IcyTree to view tree log files and maybe switching to FigTree to visualize summary trees as discussed in the next section. (Also, IcyTree can be used to export individual trees from a large log file for subsequent viewing using FigTree.) IcyTree is a tree viewer that runs in a web browser. It runs best under recent versions of Google Chrome and Mozilla Firefox (in that order).
To view MultiTypeTree log files using IcyTree, simply navigate to the IcyTree web page, select “Load from file” from the “File” menu, then select one of the tree log files using the file selection dialog. Once the file is loaded you will see the first tree it contains. In order to select a different tree, move the mouse pointer over the box in the lower-left corner of the window. This box will expand to a small dialog containing buttons allowing you to navigate between trees. The ‘<’ and ‘>’ buttons move in steps of 1 tree, while ‘<<’ and ‘>>’ move 10% of the tree file per click. You can also directly enter the index of a tree. (Note that there are keyboard shortcuts for almost all commands in IcyTree and that these can be found by selecting “Keyboard shortcuts” from the “Help” menu.)
Initially the trees edges will be uncoloured. To colour the edges according to the edge type, open the “Style” menu, navigate to the “Colour edges by” submenu and select “type”. A legend and axis can be added by choosing “Display legend” and “Display axis” from the same menu.
The following shows the final tree of h3n2.h3n2.map.log in IcyTree, which represents our sampled estimate of the MAP multi-type tree: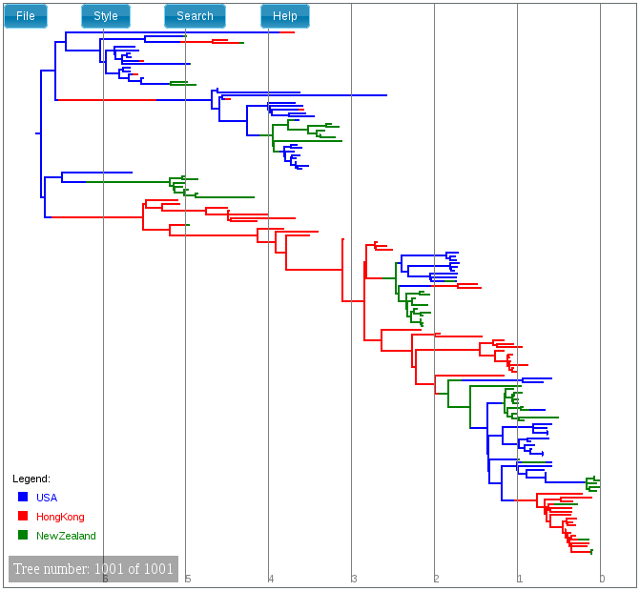
While IcyTree is useful for rapidly visualizing the results of an analysis, it is not nearly as feature-rich as FigTree and not as capable for producing publication-quality graphics. Happily, however, IcyTree can extract single trees from larger log files. Simply navigate to the desired tree, open the “File” menu, choose the “Export tree as” submenu and select “NEXUS file”. (It is important to select “NEXUS” instead of “Newick” as the Newick format does not support the annotations that MultiTypeTree uses to mark the edge types.
Producing a summary tree using TreeAnnotator
While it is tempting to view the MAP tree shown above as the primary result of the phylogenetic side of our analysis it is very important to remember that this is only a point estimate and says nothing about the uncertainty present in the result. This is an important drawback, as we have done a full Bayesian analysis and have access to a large number of samples from the full posterior in the tree log files. The MAP tree discards almost all of this information.
We can make better use of our raw analysis results by using the TreeAnnotator program which is distributed with BEAST to analyze the h3n2.h3n2.typedNode.trees which were produced by our MCMC run. To do this, simply load TreeAnnotator and select the h3n2.h3n2.typedNode.trees file as the input file and h3n2.h3n2.summary.tree as the output file. Select “Mean heights” from the “Node heights” menu and set the burn-in percentage to 10: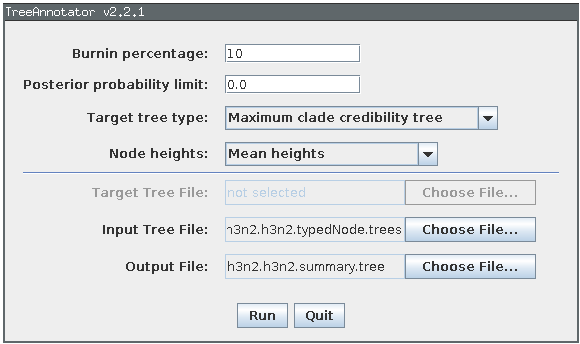
Pressing the “Run” button will now produce an annotated summary tree. To visualize this tree, start FigTree, choose File->Open, then select the file h3n2.h3n2.summary.tree using the file selection dialog. In the “Appearance” pane select “type” from the “Colour by” menu. Increase the line weight to at least 2, then select “type.prob” from the “Width by” menu. Turn on “Node bars” and select “height_95%_HPD” from the menu in the corresponding pane. Lastly, check the “Legend” check-box and select “type” from the menu in the corresponding pane to produce something like the following: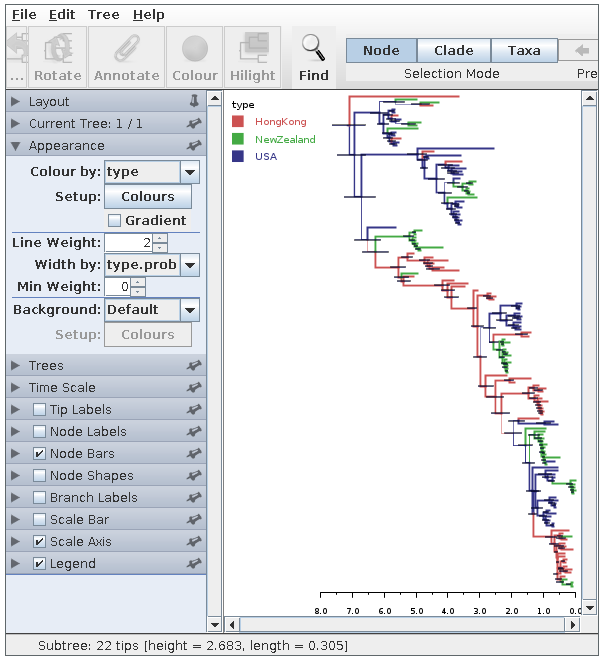
Here we have a full consensus tree annotated by the locations at coalescence nodes and showing node height uncertainty, with the widths of the edges representing how certain we can be of the location estimate at each point on the tree. This is a much more comprehensive summary of the phylogenetic side of our analysis.
Final notes
Over the course of this tutorial we have learned how to
- install the MultiTypeTree package,
- put together a structured coalescent analysis using BEAUti,
- run the analysis using BEAST, and
- process and view the results using Tracer, IcyTree, TreeAnnotator and FigTree.
Before finishing, there are three important points that need to be made. The first of these is simply this:
Estimating migration rates is hard - be prepared to wait
a long time for solid results.
This is especially true for models with 4 or more locations/types/demes. In those cases, you will probably want to start thinking about using informative priors on the rate parameters, or at very least setting strict upper (and lower) bounds on the rate parameters.
The second point is this:
Even though it’s very tempting, try not to take the MAP tree
estimate too seriously.
This is for the following reasons:
- This is only an estimate of the MAP tree.
- Even the true MAP is only a point estimate. It tells us nothing about the uncertainty associated with any of the migrations/coalescences depicted.
For quantitative purposes it is much better to look only at the marginal distributions of parameters such as those present in the parameter log file. One can also compute additional summary statistics from the tree log file using the TreeLogAnalyser utility that is distributed with BEAST.
The final point is the following:
The summary tree produced by TreeAnnotator ignores migration histories on
the tree edges.
This means that while the movement between types may seem very infrequent on the summary tree, it may in fact be extremely rapid. Thus while the summary tree is a useful picture of our analysis results, it is still important to look at the estimated MAP tree or to browse through the tree log file to get a better picture of what is going on. (Total migration event counts are also recorded in the parameter log file: posterior estimates for those counts give a much better picture of how rapidly individuals in the population are moving around.)

若有收获,就点个赞吧
0 人点赞
