- 一. 使用前介绍
- 二 . 开始使用
- (一)Open按钮导入比对后的序列文件
- (二)Options按钮修改RDP3的相关设置
- (三)各种检测方法的参数设置
- 1. RDP 检测方法的参数设置
- 2.GENECONV 检测方法的参数设置
- 3. BOOTSCAN/RECSCAN 检测方法的参数设置
- 4. MAXCHI检测方法参数设置
- 5.CHIMAERA检测方法参数设置
- 6. SISCAN检测方法参数设置
- 7.LARD检测方法参数设置
- 8. PHYLPRO Settings (一定要调)
- 9. DNA Distance Plot Settings
- 10. DSS (TOPAL) Settings (根据序列条数要进行设置)
- 11. VisRD Setting检测方法设置
- 12. Breakpoint Distribution Plot Settings
- 13. Recombination Rate Settings
- 14. Matrix Settings
- 15. Tree Settings
- 16.SCHEMA Settings
- 三.检测方法原理及结果解读
- 四. 检测方法特点速查表
背景:RDP是一款检测重组或重配的软件,适用于Windows 95/98/NT/XP/VISTA系统的软件(其实win7/8/10也可以),这款软件最大的特点是:它同时使用一系列不同的重组检测方法来检测比对后的序列中明显的重组事件,而不需要用户事先指示非重组的参考序列。但是根据用户的实际使用中发现,这个软件对于大型数据集,可能会得到许多假阳性结果。所以对于大量数据(条数比较多的序列)可以先用RDP进行疑似重组初次筛选,再将所得结果用另外一个软件Simplot进行精确的第二次重组筛选和重组断点定位,如此便可精确定位到重组。
下面是RDP3(RDP版本3)使用说明。(注:所有的截图展示的参数是程序默认值,并非本人的建议值)
参考文献:《RDP3 Instruction Manual》
一. 使用前介绍
软件RDP3可以分析细菌或者病毒序列,但是用于RDP3分析的序列是比对后对齐的序列,且RDP3不提供序列比对功能。
RDP3可以识别PHYLIP, GDE, FASTA, CLUSTAL, GCG, NEXUS, MEGA and DNAMAN格式的文件。
它包含8种检测方法,BOOTscanning,GENECONV,Maximum Chi Square,CHIMAERA,Sister Scanning, 3SEQ, VisRD, TOPAL DSS方法。
二 . 开始使用
(一)Open按钮导入比对后的序列文件
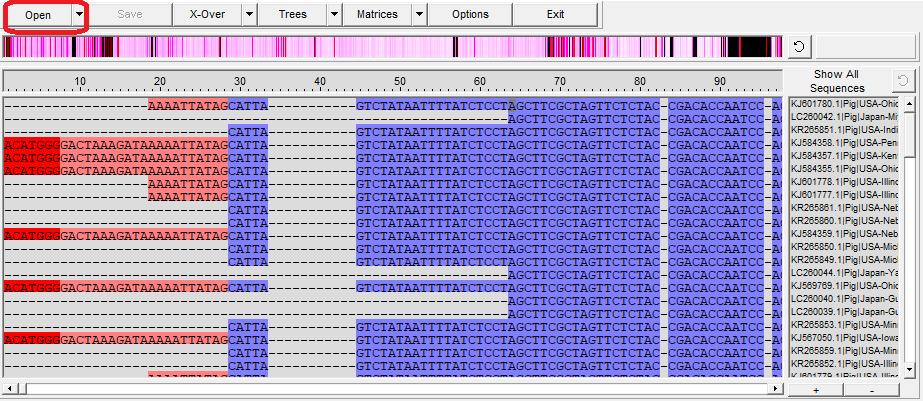 (图1)
(图1)
(二)Options按钮修改RDP3的相关设置
**点击Options按钮,会弹出下面窗口:**<br />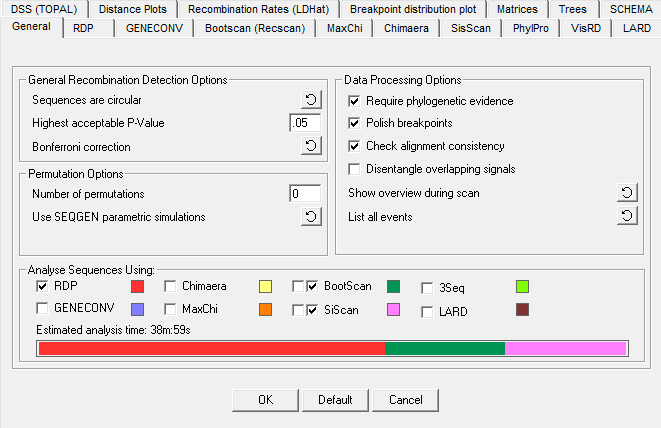<br /> **(图2)**<br />**注意:****对于RDP3的临时用户,程序的默认设置应该适用于大多数的数据集。** 需要唯一改变的设置是图中的斜体部分,但改动通常(除非真的知道怎么去改,否则改动需谨慎)只包括这些:(1)自动重组分析的方法选择;(2)具体各种方法的相关参数设置;(3)树的设置(可以在其中更改替换模型和引导重复);(4)重组率的设置。
1.General设置
①序列设置为linear 或 circular__ ,默认值是circular。
② highest acceptable p-value的设置,最高可接受的p值设置是指由于偶然机会,序列在潜在重组区域表现出高的相似性的最高可接受概率,p值的计算因不同的方法而不同。
最佳的最高p值设置取决于被分析的比对中的序列数,用于检查对齐的方法,使用的滑动窗口的大小(对于RDP,Bootstcan,MAXCHI,CHIMAERA和SISCAN),以及是否打开或关闭多重比较校正设置。
③多重比对校正设置默认是打开的,因为这样可以对于概率估计来说,是计算全局范围的p值的而不是当前选择的局部范围p值。注意,有两种多重比对校正方法可以选择, 默认值为“Bonferroni correction”,另外一个为“step-down correction”,点击右侧按钮可更改。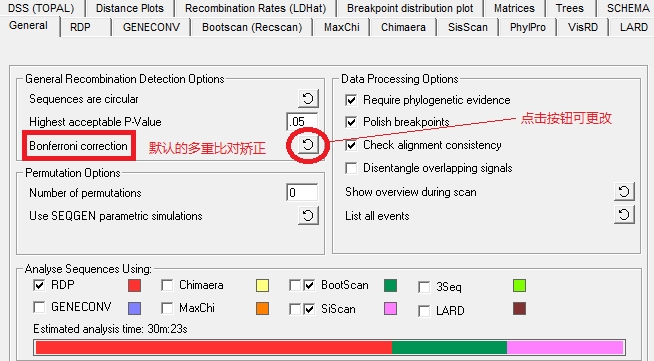
(图3)
注意:如果关闭多重比对校正,即使p值设置为0.05结果也可能会出现很多假阳性;实在要关闭多重比对校正,建议将P值下调为0.0001。
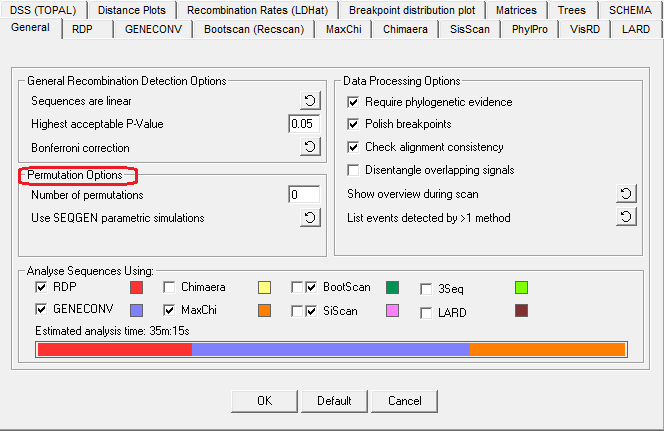
2. Permutation options
(图4)
①如果“number of permutations ”设置为大于0的数字,则RDP3将以置换模式运行其自动重组检测分析。
②RDP3可以使用两种不同的方法来模拟置换测试中使用的序列,最简单的是“shuffling alignment columns” 方法,这种方法保持序列中序列的大部分特性(例如它们的系统发育相关性和核苷酸组成),但是会破坏对齐中明显的大多数重组信号。
③同时,“shuffling alignment columns”更容易检测比对的部分中的重组,但会产生很多假阳性结果,于是第二种方法“SEQGEN”为了解决这个问题诞生了(默认的方法)。使用置换设置时要非常小心, 除了程序运行非常缓慢,它也可能意外崩溃。
3. Data processing options
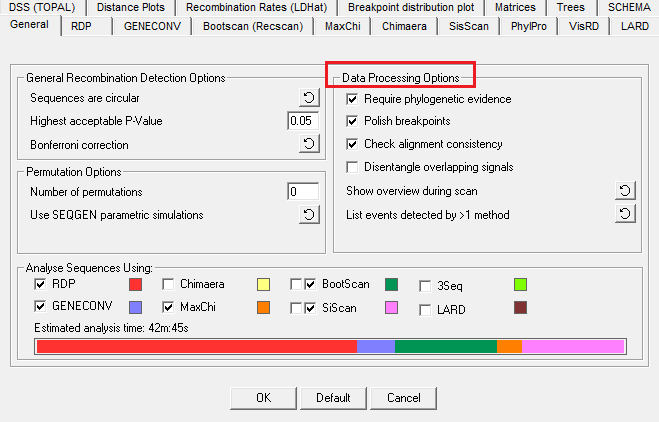
(图5)
一旦RDP3扫描了序列并列举了所有可检测的重组信号,它就开始尝试将所有可检测的重组信号提取到可以解释信号的最小组唯一重组事件(通常非常耗时)。 如果您希望了解程序运行得到的结果,则了解此过程是必要的,因为使用对齐中的多个序列组合几乎总是可检测到单个实际重组事件。
①“require phylogenetic evidence” 设置允许您指定是否希望程序没有丢弃系统发育支持的重组信号,一般我们在这里要打勾。
②在自动扫描期间,采用不同的检测方法鉴定序列重组的区域。 这些区域的边界(称为断点)通常显然不是最理想的,选择“polish breakpoints”设置将促使RDP3在所识别的那些区域附近寻找更好的断点。 即使使用此设置,您也应该意识到程序仍然可能识别的是错误的断点位置。这里也一般打勾。
③序列的错位是错误重组信号的主要原因。第三项一般也打勾。
④这个“disentangle recombination signals”设置仅应用于相对罕见数据集的重组。 如果它用于复杂数据集,其中大多数序列是重组的,只要它找不到一组亲本序列之间彼此是非重组的事件,程序就会陷入永无止境的循环分析。 您还应该意识到,没有自然的办法阻止重组序列彼此重组(即一些重组体的实际亲本序列实际上也可能是重组体)。
⑤当使用多于一种重组检测方法来扫描比对后的序列时,可以改变“list events”设置,使得RDP3仅显示大于一定数量的方法检测到的重组事件。 例如,如果你一共使用了6种不同方法进行重组检测,然后在这里选择了“> 2 method”设置,则RDP3将仅显示通过3~6不同的方法共同确认得到的重组结果。如果在分析完成后,想要放宽此设置或使其更加严格,可以来调节这里,分析结果会自动改变更新。
4.Analyse sequences using
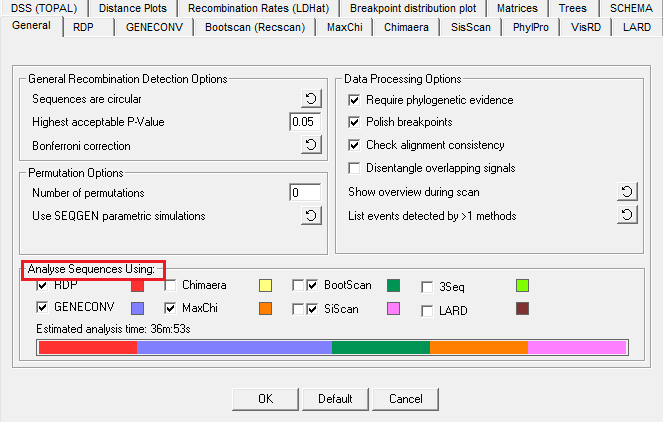
(图6)
RDP3允许您使用七种不同的重组检测方法自动分析序列。可以单独使用不同的方法或不同组合的方法。程序给出了不同方法的相对执行时间和总执行时间估计的指标。
请注意:(1)相对和总执行时间的估计可能不准确;不同的方法可能有非常不同的速度;
(2)BOOTSCAN和SISCAN有两个相关的选择框。如果选择左侧框,则将使用这些方法来探索新的重组信号。如果选择了右侧框,则该方法仅用于检查其中通过已经选择的其他“主扫描”方法检测到重组信号的序列,这种“二级”扫描模式也可采用LARD方法。在二次扫描模式中选择这些方法的原因是它们比其他检测重组信号的方法慢得多。因此,在分析大型数据集时,通常需要先使用快速方法去探索重组信号,然后再使用较慢的方法来验证这些结果。无论是否选择3SEQ,RDP,GENECONV,MAXCHI或CHIMAERA方法进行主扫描,它们总是会全部用于其他方法检测到的重组信号的二次扫描中。
(三)各种检测方法的参数设置
1. RDP 检测方法的参数设置
点击“RDP”按钮,进入RDP参数设置界面。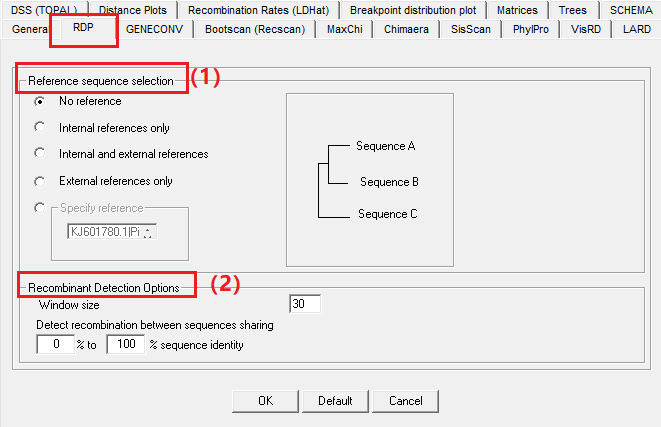
(图7)
(1)Reference sequence selection
用于在分析期间鉴定系统发育信息位点的参考序列可以以5种不同方式选择。
①默认设置为“use no reference”,这意味着无论它们是否具有系统发育信息,将检查所有位点。虽然这种设置为重组检测提供了最大的效率,但是如果检查一些非常不相似的序列(即,如果在这些序列中存在同一性小于60%的序列),它倾向于识别一些假阳性信号。如果你是通过多种方法将检测到的重组信号被接受作为重组的依据,那么在这里设置为“use no reference”是没有问题的。
②但是,如果你只是单独使用RDP这一种方法来检测重组。**(很少只用一种方法)**
你的序列里既有非常相似也有高度分歧的。(i)如果是大数据集(序列条数大于30个),使用“internal references only”设置可提供最佳的重组明确无误的估计断点;(ii)如果是小数据集(<30个序列),建议使用“internal and external references”设置。(iii)如果是非常小的数据集(<5个序列),只要数据集中的所有序列同一性大于 70%,始终建议使用“use no reference”设置。
如果你的序列包含一组密切相关的序列的数据集,并且你可以得到亲缘关系不太远的外群序列,那么外群序列可用于“specify a reference”。但是,不建议使用此设置。请注意,虽然“internal and external references”设置对小型数据集有意义,但随着数据集变大,使用此设置分析得到的结果将开始接近“no reference”设置。如果需要准确识别断点,则不建议使用“external references only”或“specify reference”设置。
(2)**Recombination detection options.
①可以设置扫描序列时使用的窗口大小。注意,RDP方法一次仅检查取样的三元组序列(即三个序列的组合)的多态性位点,注意:这种检测方法的窗口大小指的是每个窗口中包括的这些多态位点的数量。 虽然较大的窗口尺寸能降低干扰但也会降低分析的灵敏度;较小的窗口尺寸会增加灵敏度但也会增加误报的可能性。
②当分析发散序列时,一些参考序列设置可能导致高于期望的假阳性率,所以还需要将RDP分析限制为在一定范围内的同一性序列。 例如,在某一个属内,需要对种间重组**进行分析。 如果已经确定病毒种的成员具有超过90%的同一性,而属的成员具有超过80%的同一性。如果将“detect recombination between sequences sharing”设置为80和90,则表示仅检测这个属内的种间重组。
2.GENECONV 检测方法的参数设置
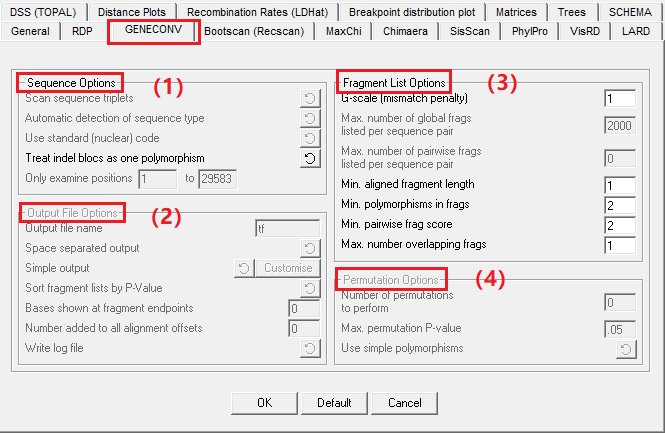
(图8)
(1)Sequence options.
①在RDP2中,可以将GENECONV设置为每次以pairs(两条)或triplets(三条)来筛选比对后的序列。在RDP3中,GENECONV方法只有triplet扫描可以用,而“scan sequence pairs”设置只能在手动的重组检测过程中使用。
②当使用“scan sequence pairs”设置时,GENECONV将对齐序列中两条序列的变异位点识别为多态性位点,然后检查每个可能重组的序列对。如果选择“scan sequence triplets”设置,程序将会把对齐中的每个可能重组的三条序列视为独立对齐,并像使用“scan sequence pairs”设置那样对它们进行筛选。因为在比对后的序列中,sequence triplets 比sequence pairs存在更多可能的重组,所以设置序列三元组体将具有更严格的多重比对校正。
③虽然GENECONV会自动检测加载的序列是DNA还是蛋白质,但可以强制将序列解释为DNA,蛋白质或DNA编码区域。如果选择“sequences are DNA coding region”设置,则可以使用标准核苷酸密码子或哺乳动物线粒体密码子来确定核苷酸取代是同义的还是非同义的。
④处理gaps(indels:“ - ”或“。”)。核苷酸序列中的相对应的一组连续的“ - ”或“。”称为gaps(gap块),可以被视为单个多态性(single polymorphism),每个单独的插入可以作为单独的多态性来对待(individual polymorphism)。使用“treat indel blocks as one polymorphism”可以将每个gap块视为单个多态性,或者可以使用“Ignore indels”简单地忽略gaps(indels),即使忽略gaps,程序的执行方式也会类似于将gaps的视为单个多态性。
最佳设置取决于你序列的对齐方式。如果比对后的序列各部分稍微偏离并且比对过程插入了大量的gaps,则最好将gaps视为单个多态性。
另外一个设置“treat each indel site as an individual polymorphism”增加了多态性位点的数量,多态性数量的增加能够鉴定更难以检测的重组区域。 但是Stanley Sawyer(GENECONV的作者)建议 “treat each indel site as an individual polymorphism”这个设置永远不要使用。
(2)Output file options.
这部分比较简单,忽略。
(3)Fragment list options.
① G-scale设置影响GENECONV如何处理错配的核苷酸。设置为0表示不允许片段内有错配,将G-scale设置为0是设置无限高的错配罚分的一种特殊情况,但是,G-scale设为1是设置最低的错配罚分。 将G-scale增加到1以上会增加错配罚分(在非常高的值处与G-scale设置为0时的错配罚分是接近的)。
没有最佳的G标度设置,应根据数据集进行调整,为了检测最近的重组事件,G-scale设置为0或者高值(5+)可能是最合适的。为了检测较旧的重组事件,G-scale值设置1或2可能是最佳的。默认值是1。
②“The maximum number of fragments” 只有在查找最可能的重组事件时,才能更改显示的最大片段数。
③在程序执行期间,geneconv可以设置忽略某些潜在的重组区域,有以下几种情况:
(a)小于一定长度; (b) polymorphic sites的意思是少于一定数量的多态性位点(该设置对于区分序列保守和重组是有用的);(c)pair-wise score意思是低于一个特定截止点的pair-wise得分;(d)overlapping frags设置:设置与较低p值片段重叠的较高p值片段数量,默认是1。
(4)Permutation Options
①GENECONV可以使用置换测试来检验潜在重组区域的显著性。
② number of permutations(排列)设置,排列数要和分析的长度成正比,p值设置为0.05对于1000个数的permutations可以防止检测到假阳性率。
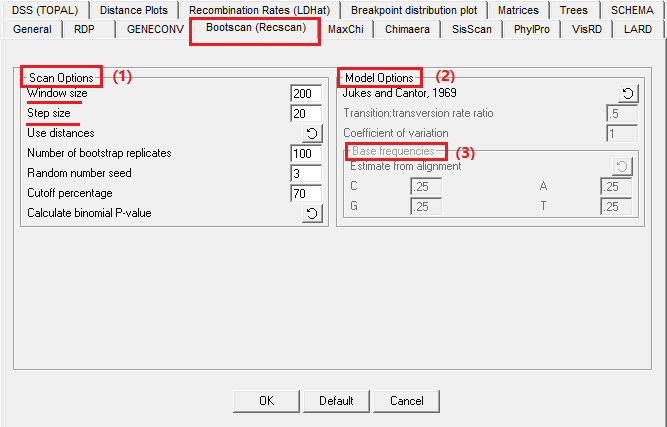
3. BOOTSCAN/RECSCAN 检测方法的参数设置
(图9)
(1)Scan options
①这个“window sizes”值的设置很重要,它的意思是,对于我们整条序列,BOOTSCAN每次分析的片段长度,默认的BOOTSCAN值是200, 也就是每次分析200个bp的片段(会同时扫描变异位点和不变位点)。**同时,这个值的大小与我们预期的重组区域的大小是相关的。(理解这句话的含义)
②step size值必须小于window sizes值,推荐小于window sizes值的50%。window sizes值越大可以增加检测信号,如果一个真实存在的重组片段的大小仅仅略小于你设定的window sizes值,那么这个重组片段几乎是检测不到的。
③如果用Use distances这个选项,分析速度会快些,可以点击右边的“回旋形状”的按钮切换到使用进化树的模式,如“Use UPGMA trees” 或者 “Use Neighbour Joining trees”。软件作者建议最好不要使用UPGMA trees,而是去使用Neighbour Joining trees或者distances。
除非你的序列里的不同区域有着非常不同的进化速率,一般来说Use distances和Use Neighbour Joining trees及Use UPGMA trees得到的结果是一致的。默认值是Use distances。
④bootstrap replicates值的设置,也就是自举重复数(自展值或叫自举值)的设置。**软件作者强烈建议对于包含或20个 2Kb以上的序列数据集,自举重复数保持在1000以下,可以通过增加cutoff percentage比来控制结果的显著性。一般而言,使用200的自举重复数同时采取95%的cutoff,与其他方法中使用0.05 p-value cutoff同时使用多重比对校正,获得的结果是类似的。
⑤什么是cutoff percentage ?
这里的cutoff percentage指的是在三条序列中有潜在重组的bootstarp support,说得通俗点就是bootstrap占多少比例才有可能是潜在的重组,比如默认是设置70,也就是bootstrap占比70%时才判断为是潜在的重组。需要注意的是BOOTSCAN检测方法里的95%的cutoff和其他检测方法里的p-value cutoff of 5%(i.e. 0.05)不是同一个东西,不要搞混淆。BOOTSCAN检测方法里的95%的cutoff值的是指当bootstrap占比95%时表示重组事件显著,而其他检测方法里的p-value cutoff of 5%说的是置信域。
毋庸置疑,使用更多的bootstrap replicates和更高的cutoff percentage会使检测到的结果更加可靠。
⑥Random number seed:这个用默认值就行。
⑦虽然对于BOOTSCAN检测方法来说,可以简单地将bootstrap values看做P-value,但是软件作者建议使用“calculate binomial p-value”或者“calculate Chi Square p-value”,默认是“calculate binomial p-value”,因为软件作者发现“calculate binomial p-value”是最强大的设置。
(2)Model options
①如图9所示,默认的模型是Jukes-Cantor 1969 模型,另外还有其他几个模型可供选择。
②当四个碱基的频率相等时,Kimura 模型和 Felsenstein, 1984 模型(F84)是相同的。F84模型容许在均衡基础频率上存在差异,Jin-Nei, 1990模型与模型很相似。但是Jin-Nei, 1990模型假设在不同的位点发生不同的替代率,使用的是GAMMA速率,shape值由变异系数(coefficient of variation)确定,低值异味着不同位点以相似的速率进化,高值意味着不同位点进化速率差异大。RDP3里是使用phylip的DNADIST的组件来计算遗传距离。
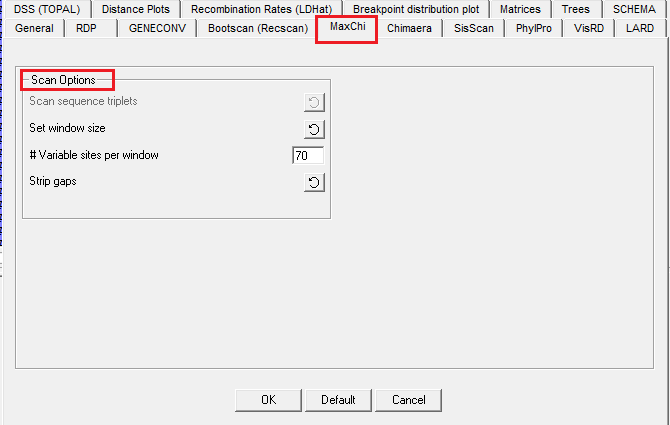
4. MAXCHI检测方法参数设置
(图10)
(1)Scan options
①在RDP3中,在自动重组检测期间每次只能扫描三条序列(三元组扫描,下同)的变异位点。(RDP, CHIMAERA, GENECONV and 3SEQ methods, MAXCHI only examines variable nucleotide position。) 但是,当使用MAXCHI手动筛选序列以检测重组时,仍然可以进行二元组扫描(每次扫描两条序列,下同)。 三元组和二元组扫描之间的主要差异在于二元组扫描不能正确鉴定亲本和重组序列。
②set window size 当选择的为set window size时,与之对应的是variable sites per window, 用于检测具有20个变异核苷酸位点的重组区域的最佳窗口大小是40。
③选择“scan triplets”设置后,最好再设置variable window size,点击“set window size”右侧的按钮切换,如下图: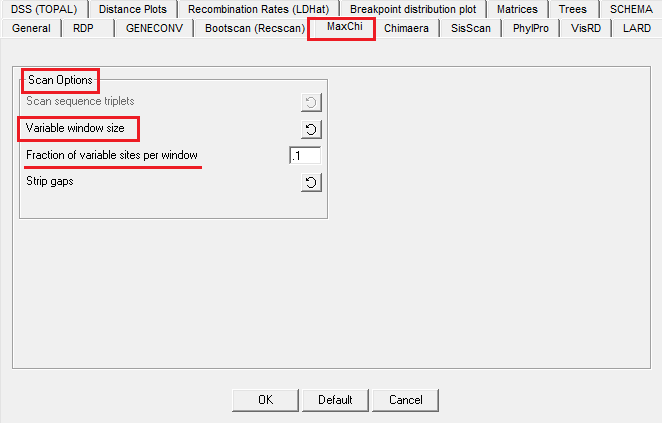
(图11)
在这里可以指定每个窗口变异位点的比例(图中画横线部分),使用variable window size的特点是,当三元组中包含较多差异大的序列时,窗口会变大;三元组中包含较高度相似的序列时,窗口会变小。注意,如果三条序列的总变异位点数少于窗口设定值的1.5倍,则窗口设定值将自动调为总变异位点数的0.75倍。如果设定值小于10,则不扫描检测。
5.CHIMAERA检测方法参数设置
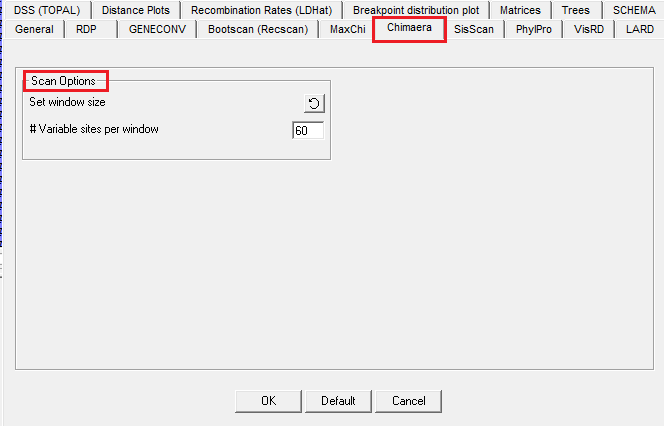
(图12)
需要注意的是:GENECONV,3SEQ、MAXCHI和CHIMAERA仅检查可变核苷酸位置,所以CHIMAERA里的窗口大小是指可变位点的数量而不是总核苷酸的数量。用于检测具有20个可变核苷酸位点的重组区域的最佳窗口大小是40。同样的注意,如果三条序列的总变异位点数少于窗口设定值的1.5倍,则窗口设定值将自动调为总变异位点数的0.75倍。如果设定值小于10,则不扫描检测。
6. SISCAN检测方法参数设置
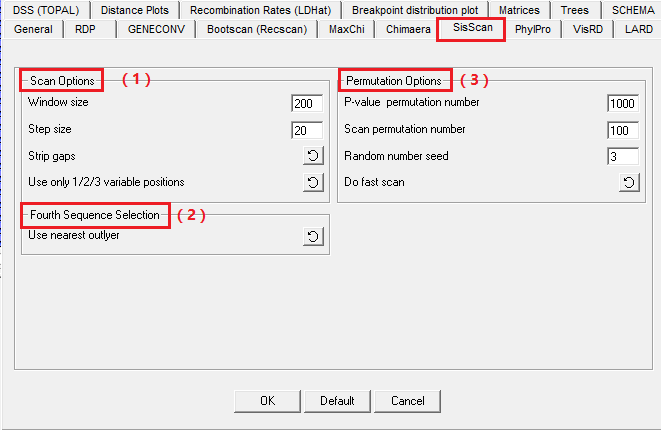
(图13)
(1)Scan options
①这个检测方法里的window size是指每次分析的片段长度,而step size要求小于window size的50%。
②strip gaps(去掉空位)。软件作者强烈建议设置为去掉空位,如果使用空位(-或者.),则空位会被看做第5种碱基。
③软件作者强烈建议设置为“use only 1/2/3 variable positions”。如果设置为“use only 1/2/3/4 variable positions”将重点分析三元组序列中的差异,设置为“use all positions”会检查所有可变和不变的位点。建议使用“use only 1/2/3 variable positions”设置,因为其他设置在分析中倾向于包含许多不相关的位点来“减弱”重组信号。
(2)Fourth sequence selection
SISCAN检测三元组序列(三条序列)的潜在重组时,会附带上第4条外群序列一起来分析。这条外群可以是这三元组序列外的任意一条,也可以是随机生成的。如果设置为“use nearest outlie”,RDP3会扫描离三元组序列(三条序列)最近的那一条用作外群;如果设置为“use most divergent sequence”,三元组会使用所有序列中最分散的那条作为外群;如果设置为“use randomised sequence”,对于每个窗口里用来分析的三元组序列,使用一条新的随机序列。建议设置为“use nearest outlie”
(3)Permutation options
①** scan permutation number 探索性阶段扫描先可以使用较少的数量,等到发现有潜在重组时,再使用更多数量的scan permutations。
② random number seed 在重复分析中使用相同的随机数将确保SISCAN结果是可重复的。
③另外还可以选择“do fast scan”或者“do slow exhaustive scan”。**
7.LARD检测方法参数设置
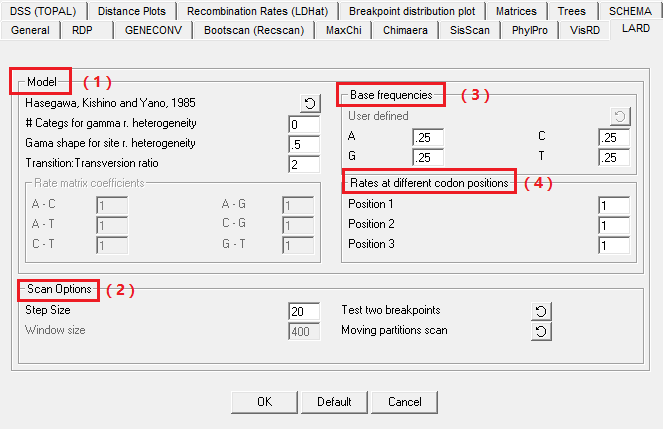
(图14)
(1)Model options.
①LADR提供了HKY、F84、、三种不同的核苷酸替代模型,使用最大似然法重建三条序列的系统发育关系。HKY允许不同的转换和颠换率以及不等的核苷酸频率,F84与HKY类似,但是F84允许从序列中估计核苷酸频率,可以以不同的方式处理转换/颠换率。
②除了核苷酸替代模型,还提供了GAMMA变异速率模型和GAMMA的shape设定。(可以先用一些模型软件测出GAMMA的shape值)
(2)Scan options
①需要注意的是,window size表示每次用来分析的三条序列的核苷酸位点总数(包含变异位点和不变位点),窗口会平均分成两个区(这个与MAXCHI和CHIMAERA以及下面的PHYLPRO检测方法有点类似),因此这里的窗口大小设置为400表示的是窗口左侧有200个核苷酸与右侧有200个核苷酸。 用LARD方法检查保守和可变序列的位置,窗口大小设置应足够大,。LARD的窗口大小应该是BOOTSCAN和SISCAN方法推荐大小的两倍。
②step size 设置为10应该是一个很好的折衷方案.
这里可以由一些模型软件测出,比如modeltest,jmodeltest等等。
8. PHYLPRO Settings (一定要调)
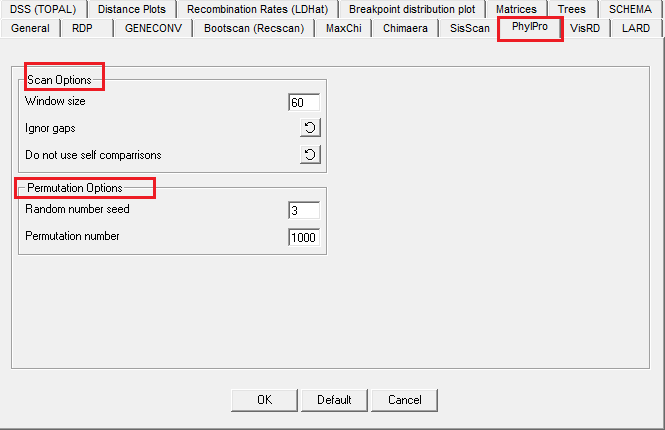
有关PHYLPRO设置和PHYLPRO工作原理的更多信息,请参阅8.8或PHYLPRO手册。 它可以从以下网址下载:
http://www.rsbs.anu.edu.au/ResearchGroups/GIG/Products/phylpro/
(1)Scan options
①PHYLPRO是另一种重组检测方法(和LARD,BOOTSCAN和SISCAN方法相似),它会检查序列的可变和保守的位点。 窗口大小设置应足够大去包含20个或更多可变位点。PHYLPRO的窗口大小应该是BOOTSCAN和SISCAN方法推荐大小的两倍,因为PHYLPRO方法涉及沿着对齐长度移动一个带有分区的窗口,窗口的每一半都与另一半进行比较。window size表示每个扫描窗口包含的变异位点数量,默认值是60。
②可以设置PHYLPRO方法以两种不同的方式处理gap:可以完全忽略任何gap,或者可以考虑这些gap。
9. DNA Distance Plot Settings
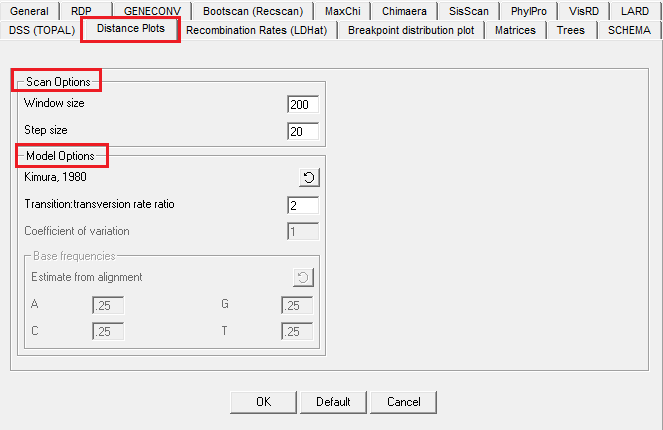
(1)Scan options
根据用于检测的亲本的相关性来设置窗口大小。每个窗口应包含至少5个可变位点。 最佳步长(step size)还取决于被检查序列的相关性,并且应小于窗口大小的20%。
(2)模型选择
推荐使用默认值。
10. DSS (TOPAL) Settings (根据序列条数要进行设置)
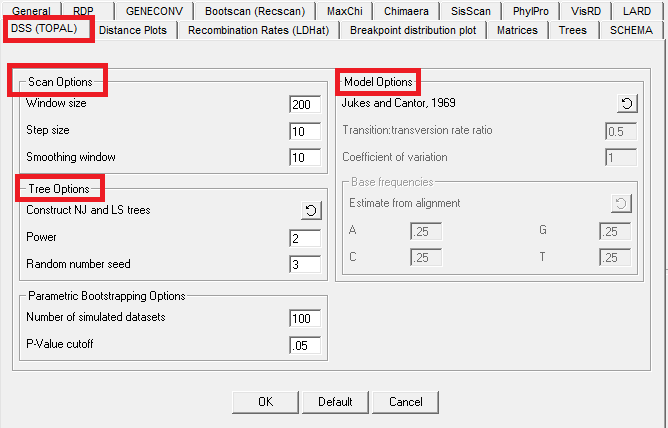
(1)Scan Options
这里需要注意的是TOPAL方法与PHYLPRO方法类似,因为窗口被分成两部分,并且其最佳的窗口大小是BOOTSCAN和SISCAN方法的两倍。 最好应该尝试设置窗口大小,以便每个窗口将覆盖超过~20个可变核苷酸位置。
(2)Tree options
①这里可以设值使用的树的类型,默认值是“construct NJ and LS trees”。“construct NJ and LS trees”这个设置比“construct only LS trees”设置运行速度快,但该方法开发人员表示,“construct NJ and LS trees”这个设置应仅在> 20个序列的手动TOPAL分析期间使用。
②“Power”设置将影响计算的DSS值的大小 , 如果DSS值非常小(例如0.002),则增加Power值会将它们增加到更容易比较的值。
(3)Parametric bootstrapping options
如果“number of permutations”超过10,RDP3将执行称为参数bootstrap测试,以确定任何检测到的DSS峰值的显著性。默认100,个人推荐200。
(4)Model options
推荐使用默认值。
11. VisRD Setting检测方法设置
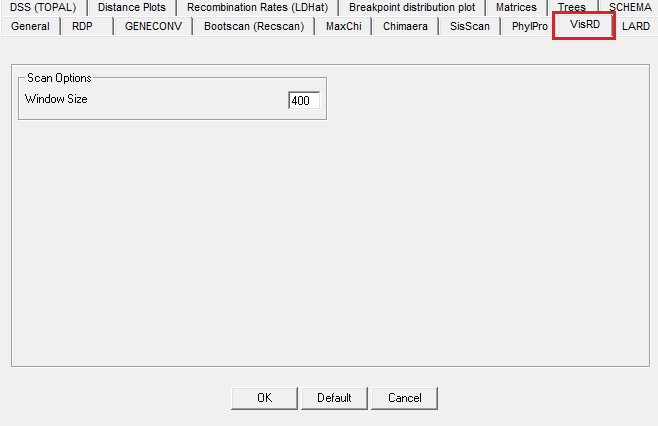
VisRD检测方法和 PHYLPRO, LARD, BOOTSCAN及SISCAN检测方法有点类似,会分析包含可变位点和不变位点,扫描窗口大小是唯一可以更改的设置,应该足够大,窗口应包含10个或更多可变位点。
12. Breakpoint Distribution Plot Settings
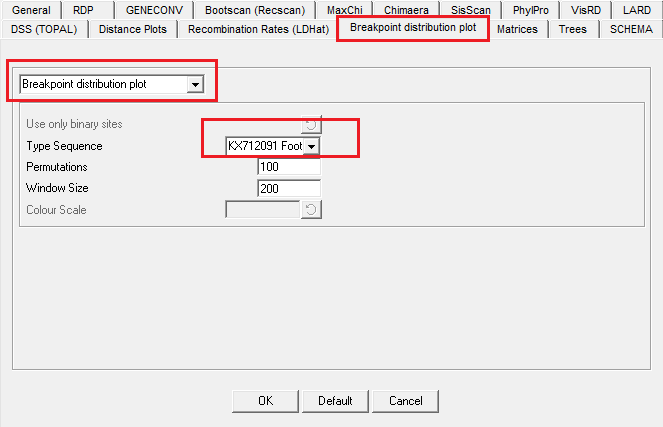
断点分布图是分析重组热点和冷点的有用方法,用于检测序列断点热点和冷点的测试, 可以指定此测试中使用的permutations数,并且应该为100或更大。 可以使用“window size””设置指定要检查的断点集群的大小。 注意,小窗口尺寸(<= 50nts)对于检测异常紧密的断点簇(即高度聚焦的重组热点)是有用的,但对于检测重组冷点或分散的重组热点不是很好。 窗口大小在100到200 nts之间通常是检测热点和冷点之间的良好折衷,但可能会错过小于指定窗口大小的区域内异常紧密的断点簇的事件。 因此,建议尝试一系列窗口大小设置。
13. Recombination Rate Settings
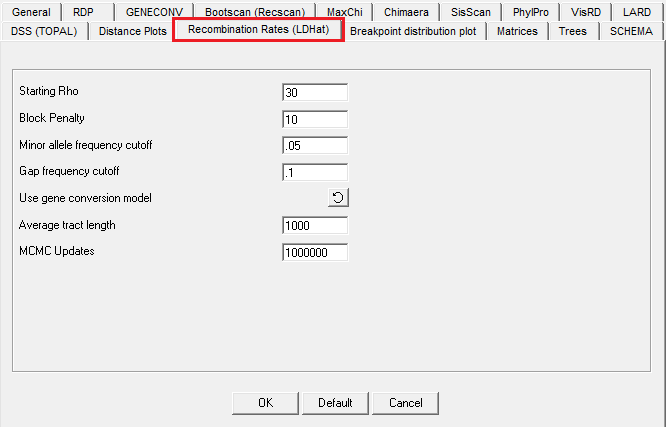
RDP3使用来自LDHAT包的程序CONVERT和INTERVAL(McVean等,2002; McVean等,2004)来构建跨序列的不同重组率的图。 有关这些程序使用的设置的其他信息,请参阅LDHAT手册:http://www.stats.ox.ac.uk/~mcvean/LDhat/instructions.html
(1)“starting Rho” 值应该是介于0和100之间的数字,理想情况下应该是对齐范围的总体缩放重组率的实际估计值。
可以先通过绘制具有任意起始rho值的图可以获得对此的估计,显示在重组信息面板中的该值可以在重做绘图时用作更好的起始值。
(2)block penalty ,指定“block penalty”以防止RJMCMC调用序列区域中重组率的太多变化 - 即可以设置block penalty以防止INTERVAL对数据过度拟合复杂变量重组率模型。软件作者推荐可以尝试构建0到50之间的一系列block penalty。较低的罚分使分析能够检测重组率中较小和更微妙的变化,但也可能导致对数据的推断变化的过度拟合。建议设置5-30。
(3)“minor allele frequency cutoff” 每次分析都需要更改变此设置的值。 例如,对于包含100个序列的比对,0.05的minor allele frequency cutoff将排除六条序列在该位置共享两个备选核苷酸之一的可变位点。
(4)“gap frequency cutoff” ,从分析中排除任何具有超过一定数量的缺失数据的序列。
(5)using and not using a gene conversion model区别不大。
(6)不同的average tract lengths值对序列中预期每个重组事件一个断点区别不大。
(7)MCMC updates ,设置MCMC代数,前10%老化样本被舍弃。小数据集100万代或者200万代够用,大数据集需要500万代或者更多代数。
14. Matrix Settings
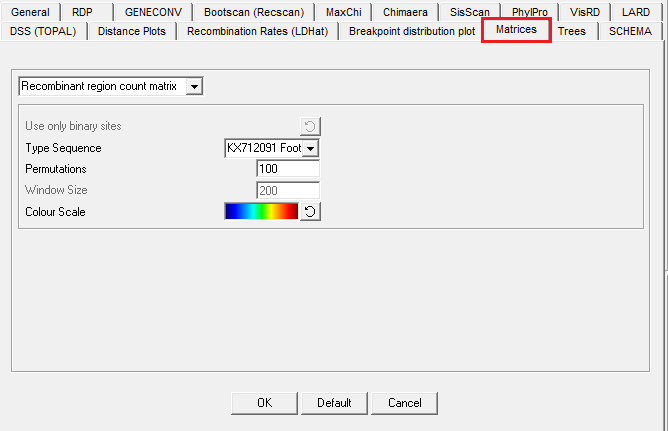
RDP3可以绘制了几种不同类型的矩阵。 许多不同的矩阵共享设置,例如颜色标度,排列数和窗口大小。①
(1) Compatibility matrix
在构造兼容性矩阵时,以各种方式处理具有两个以上字符状态的变量对齐。 通过将嘌呤和金字塔组合在一起,它们可以被忽略或“强制”成二元状态。
(2) Recombination matrix
选项“type sequence”可用于指定比对中的序列,该序列在编号绘制的核苷酸坐标编号时将用作参考。
(3)Modularity matrix
选项“type sequence”设置同上, 这里的“window size”设置是指在当检测重组体的亲本序列彼此相似程度时检查的核苷酸数。
(4)Recombination region count matrix.
选项“type sequence”设置同上,这里的“window size”指的是圆的直径,即断点对矩阵上每个重组断点对绘制的圆。
(5)Breakpoint distribution plot.
选项“type sequence”设置同上。
15. Tree Settings
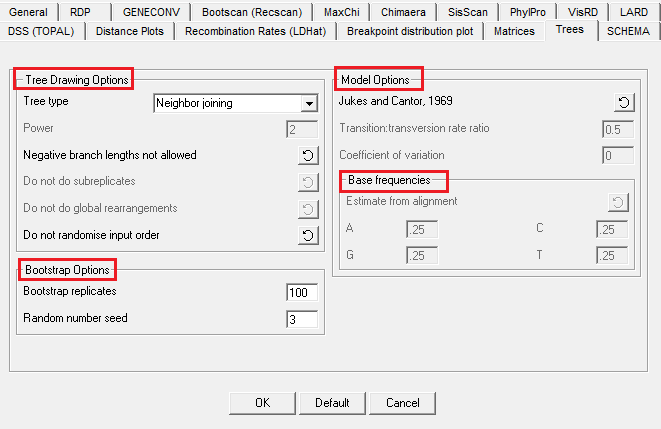
设置这个之前,希望您對不同的树,比如NJ树、BI树,ML树有很清楚的了解,这里是门大学问,一言难尽,所以省略;另外bootstarp,以及不同的model这些也是需要去了解其含义的。比如如果你选择了BI树,也会出现建BI树的一系列选项。
我希望您在使用RDP3之前已经对不同方法构建系统发育树已经非常熟悉了,如果不熟悉,就用默认的neighbor joining (NJ)树。
16.SCHEMA Settings
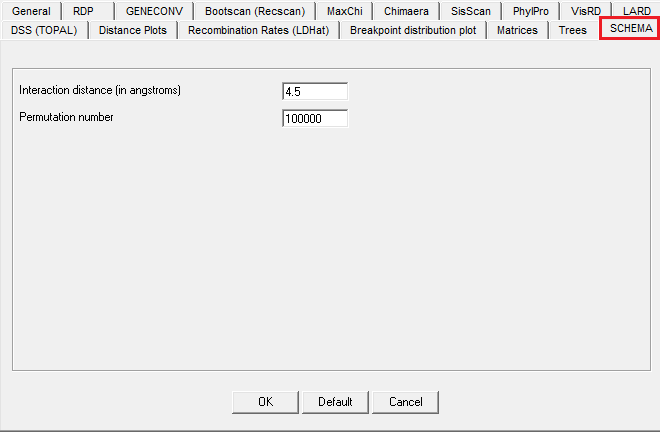
SCHEMA 是一种采用蛋白质原子坐标并估计重组蛋白中预期蛋白质折叠破坏程度的方法。 SCHEMA方法通过用户定义的彼此距离(通常在2到20 angstroms 之间)的寻找所有氨基酸对,并确定这些折叠的蛋白质内可能相互作用,可以使用“interaction distance”来定义该距离。
三.检测方法原理及结果解读
1.RDP检测方法
最初的RDP方法(Martin和Rybicki,2000)通过使用三步法检查每个三元组序列(三条序列的组合)来筛选重组事件。
(1)步骤如下
①取样三个序列,丢弃序列上所有的没有系统发育信息的位点,构建UPGMA树,确定其中亲缘最近的两条序列。无系统发育信息的位点是在指三条序列中都相同的位点,或者在三条序列中都不同的位点,或者该位点不存在于一组参考序列的任何成员中(如果使用参考序列设置)。
②沿着对齐序列的信息位点移动窗口,每次一个核苷酸,并且在每个位置计算三个配对(如A-B,B-C,C-A)中的每一个配对的indentity平均百分比。A-C或B-C的indentity百分比高于A-B的区域被鉴定为潜在的重组区域。
③使用二项分布来检验,计算多重比较校正(或Bonferroni校正的)p值。
原理图如下: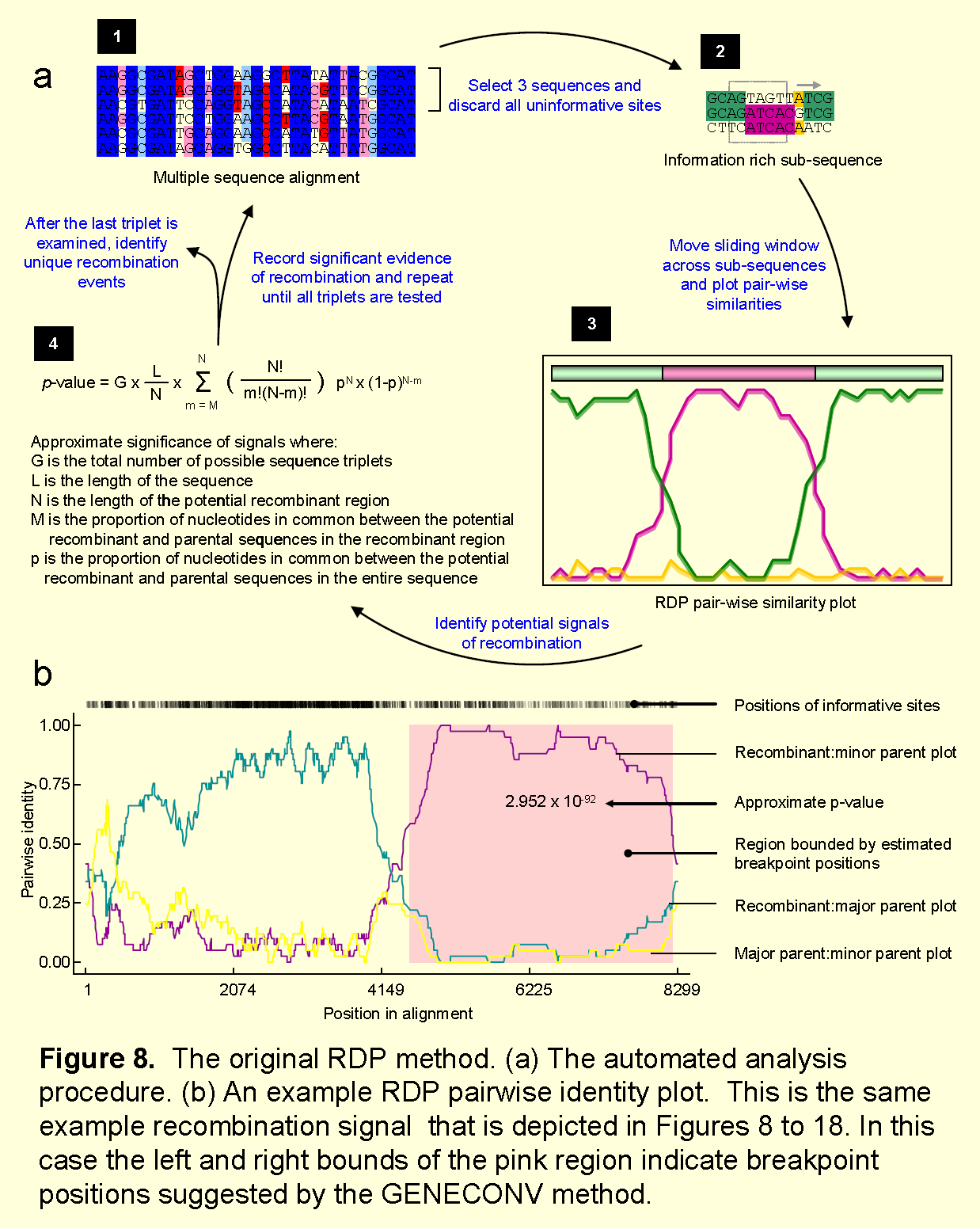
(2)潜在的问题
取决于所使用的参考序列选择方法,RDP方法可能无法分析对齐序列中的某些三元组序列的重组。例如,如果使用“use only internal references”设置,则RDP方法不会分析彼此亲缘最近的三元组序列。此外,如果一次扫描对齐序列中的4序序列,RDP方法将不能检查3种可能组合的三元组中的任何一组,除非用于建UPGMA树的序列具有适当的分支。因此,对于短序列,建议应始终使用“internal and external references” 或 the “no references”设置。
原始RDP算法无法明确处理跨谱系的速率变化(导致非超参数/非时钟状树 - 即,对齐序列中的不同序列似乎以非常不同的速率发展的树)。如果任一序列以不同的速率进化或者在不同的时间进行采样,则存在这样的可能性:依赖于UPGMA(参考序列的选择)的方法部分将不会按照预期的方式起作用。对于此类数据集,您应使用“use internal and external references” 或者 “use no references”设置(后者是默认设置)。
2.GENECONV检测方法
通过寻找序列中对齐的区域,其中“序列对”要足够相似以怀疑它们可能是通过重组产生的。 请注意,三元组扫描的方法(用于探索性分析)与pair scanning(用于手动分析)的方法相同,不同之处在于三元组扫描不是一次性分析所有的序列,而是每次将序列分成对齐的三条序列,再一一分析。
(1)步骤如下
①从对齐的序列中排除单态位点,剩下的是多态为点的对齐。
②对于序列中的每个序列对,对相同的和异常长的,或者具有异常高的相似性的“序列对”的进行评分。基于以下方案针对相似性进行评分:(a)匹配(或一致位点)计为+1,对不匹配(或叫不一致)的位点的进行罚分。 错配罚分取决于两个序列之间多态性位点的密度以及您之前GENECONV设置里在指定的mismatch intensity parameter 或者 G-scale值。
③将P值分配给高评分区域(也称为片段、高评分对齐区域或HSAP)。分配给这些区域的p值是通过Figure.9(a)排列(缓慢但准确)或(b)BLAST衍生的Karlin和Altschul(KA,1990)方法(近似但快速)得到的。虽然近似,多重比较校正(也称为Bonferroni校正或全局)KA p值通常比置换p值更保守。多重比较校正仅涉及成对的KA p值乘以在分析期间进行的成对比较的数量。
原理图如下:
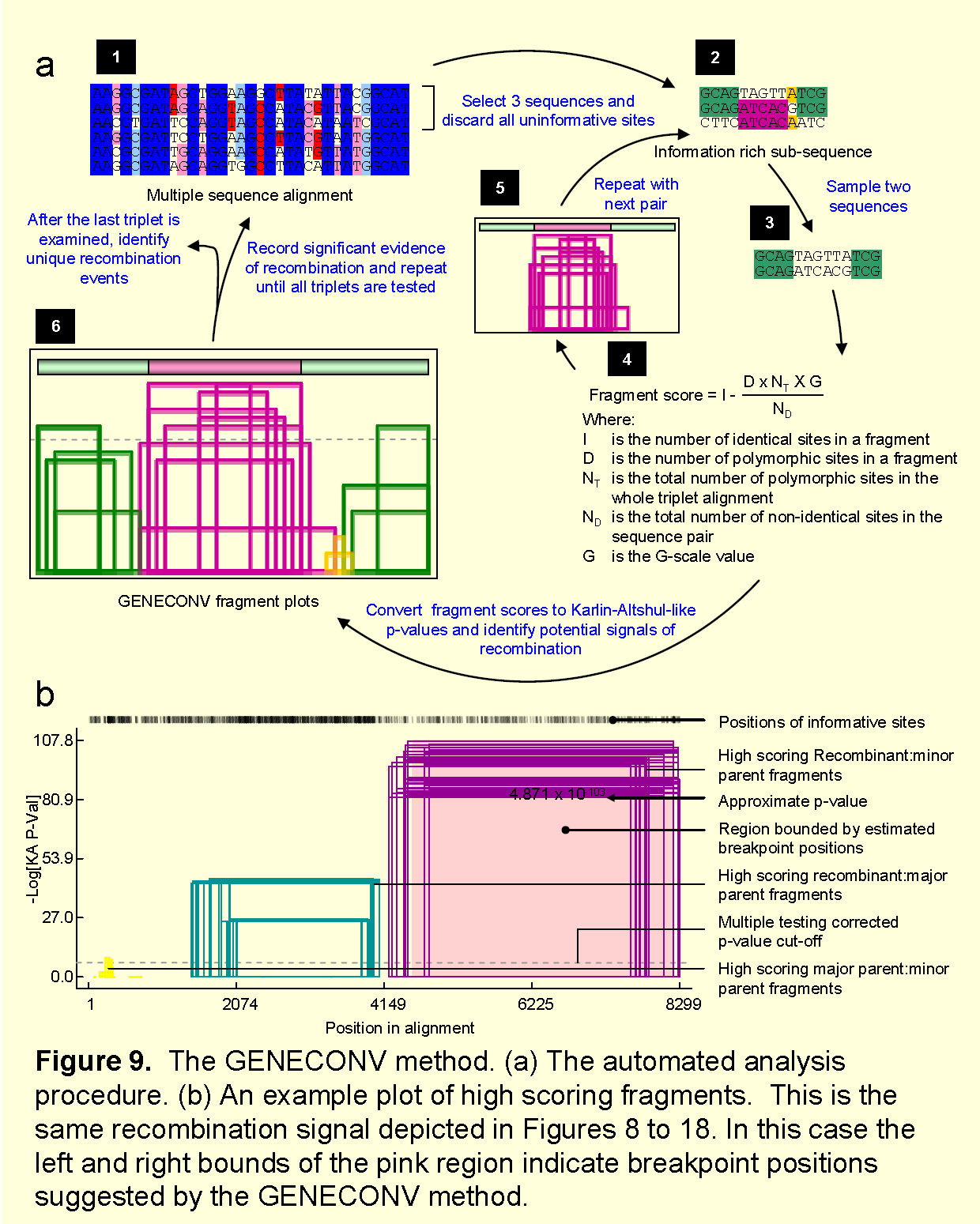
(2)潜在问题。
GENECONV适用于整个对齐序列有确定的多态位点。进行成对扫描或三元组扫描时(但特别是成对扫描时)对齐中的单个高度分歧序列将会引入许多多态性位点,这些位点与检测序列中彼此更相似的序列的重组无关。这些不相关的位点可能对分析产生两种影响:(1)当GENECONV检查密切相关的序列时,它们可能导致位点的明显一致性(这些位点将被GENECONV解释为重组区域)。 (2)不必要地增加多态性位点的数量,它们将降低p值的显著性并且可能导致错过小的(但是真实的)重组区域。因此,在使用GENECONV进行分析之前,应注意在正式分析前选择序列或在RDP3中禁用可能存在问题的序列。
如果你发现使用其他方法获得的重组结果GENECONV可以检到,但GENECONV在单独自动分析期间未检测到这些重组结果,则GENECONV很可能在数据集结构方面存在问题,执行三元组扫描代替成对扫描可以解决此问题。
在分析GENECONV的结果时,如果要接受两个序列之间发生重组,你应该始终非常谨慎,如果这两个序列是彼此最近的亲属,有可能是序列之间的一系列保守位点被误解为重组。
GENECOV的最后一个问题是它具有七种方法中最低的重组断点检测精度,对于GENECONV检测到的重组断点的位置再要通过MAXCHI和CHIMAERA方法去确认(RDP3中实现的最准确的断点检测方法)
3.BOOTSCAN/RECSCAN 检测方法
BOOTSCAN是一种滑动窗口方法,其被开发用于鉴定已知或可疑重组区域内的亲本(Salminen等人,1995)。最初BOOTSCANning涉及:(1)构建含有潜在重组序列和一组(非重组)参考序列的对齐。 (2)沿着设定长度的窗口移动一定数量的核苷酸,并为每个窗口计算构建的NJ树的bootstrap值。 (3)通过相对bootstrap法,用每个窗口的每条参考序列绘制潜在重组序列的最近邻的序列。非重组序列应该在其整个长度上只具有单一的参考序列(具有超过约70%的支持),但重组序列应该交替地(具有超过约70%的支持)与两个或更多个不同的参考序列组合。
(1)方法步骤
①设定每一次沿着对齐的序列移动指定数量的核苷酸窗口大小;
②每个窗口都进行Bootstrap replicates,使用 a pair-wise distance BOOTSCAN or they can be used in a UPGMA or neighbour joining tree BOOTSCAN计算成对序列的遗传距离。
③在每个窗口位置处,通过 bootstrap replicates确定序列中每个可能序列三元组的相对分组(基于成对距离或树位置)。核苷酸序列的距离和树都使用PHYLIP组件DNADIST和NEIGHBOR来计算。
④在完成最后一个窗口的扫描之后,扫描所有窗口上的每个三元组序列中的每两条序列bootstrap值,并扫描序列对的相bootstrap改变的地方,在两条不同序列之间交替的高bootstrap值(figure10b)的地方指示可能有潜在的重组。
⑤ 再通过二项分布或者卡方检验的p值来进行重组的验证。
原理图如下: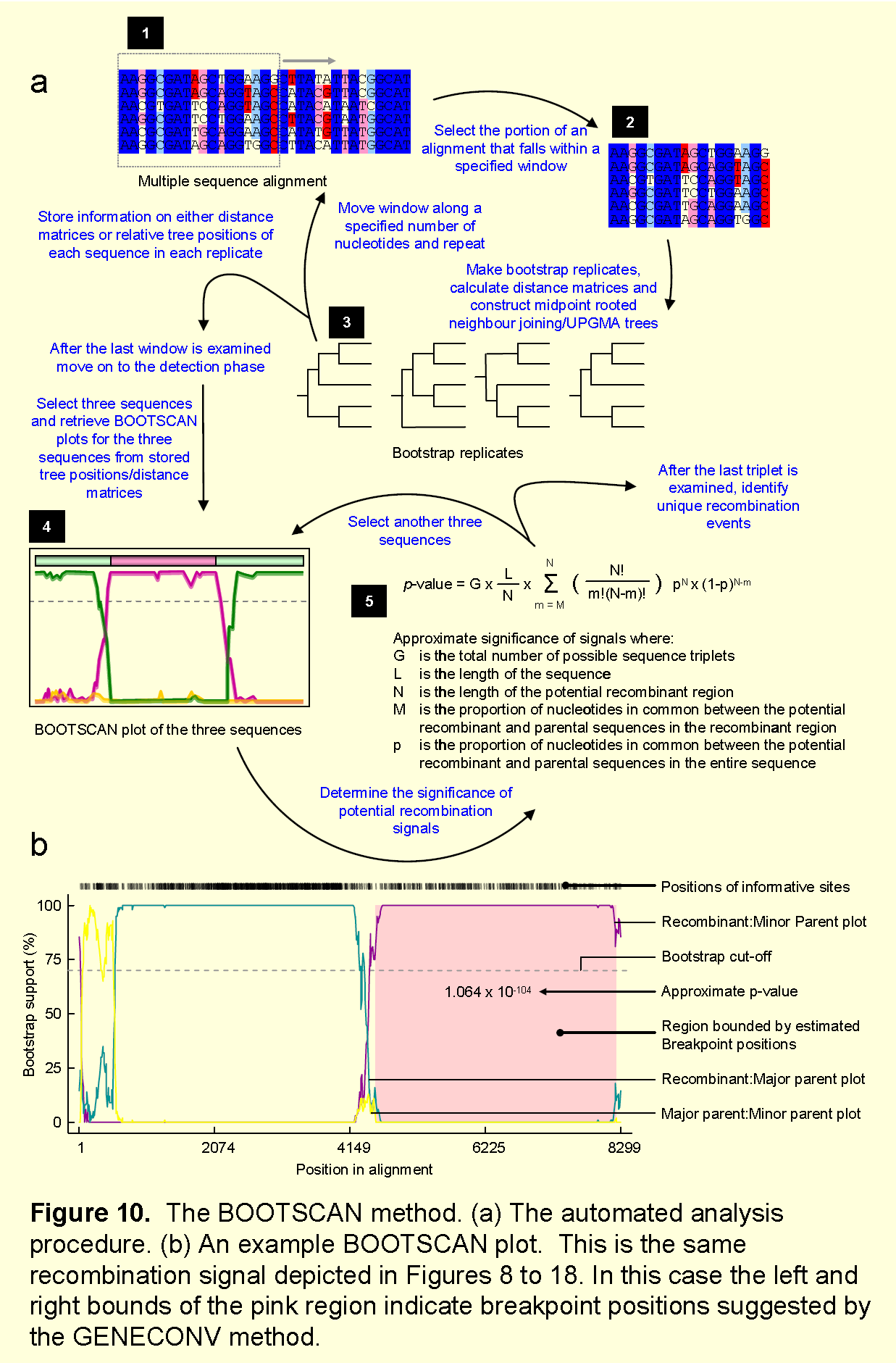
(2)潜在问题
BOOTSCAN没有定义的“适当”的水平去评价重组,比如高于某个水平的bootstrap值可以高度确信检测到的区域是重组的,广泛接受的是,95%的**bootstrap**表示更密切的相关。此外,虽然bootstrap值通常是重要的保守指标(there is therefore a good chance that many real recombinant regions will be missed with a bootstrap cutoff of, for example, 95%),没有明显的方法来纠正多个bootstrap值测试。这意味着在分析大型数据集时完全依赖bootstrap分数可能会产生大量的误报。
4.MAXCHI检测方法
Maynard Smith(1992)提出了一种方法(称为最大χ2方法),**用于识别重组断点**。MAXCHI检查序列对并通过寻找序列的相邻区域中**可变和非可变多态性位点的比例的显著性差异来识别重组断点**。<br /> **尽管比较两个亲本序列和重组序列时最大χ2方法表现最佳**,但当使用该方法检查超过3条对齐序列时,可以将RDP3设置为一次检查三条序列(“scan triplets”设置)或检查整个对齐序列中有确定的变异位点位点的两条序列(在“manual”MAXCHI扫描中实施)。<br /> MAXCHI提供了关于潜在断点的位置,但没有给出重组区域范围**。** 当使用“**scan triplets**设置时,RDP3将进行(相当粗略的)尝试匹配潜在断点,并假设断点之间的序列在单个重组区域内。)
(1)方法步骤
①对齐的序列中的所有单态位点都被丢弃,剩下的是多态位点,还可以选择是否丢弃gap位点。
②对于对齐中的每个可能的序列对,按照设定长度的窗口(在其中心具有分区)沿着序列一次移动一个核苷酸;
③在每个窗口位置处,计算2 ×χ2值用来表达分区两侧的序列对之间的可变位点数量的差异。 当沿着比对的长度绘制时,这些χ2值中的峰表示潜在的重组断裂点。
原理图如下: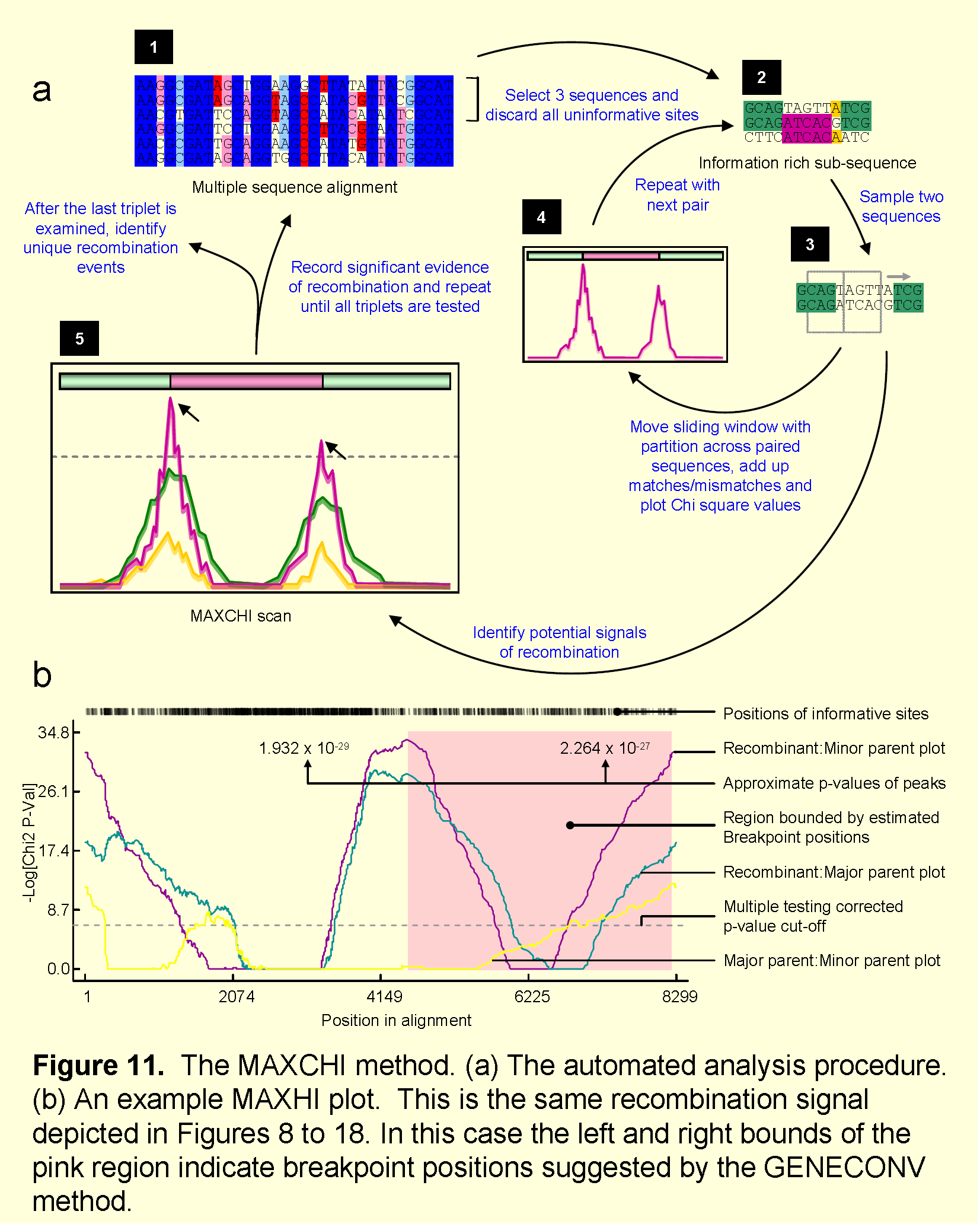
(2)潜在问题
使用MAXCHI扫描三元组序列最严重的问题是它会分析三个序列之间所有的差异位点。在这些情况下,MAXCHI将产生似乎是一些非常有说服力的重组信号,而结果可能是假阳性 - 即具有高p值的结果,其不能用任何其他重组检测方法确认。
与GENECONV一样,在手动MAXCHI分析期间对序列进行成对扫描使用来识别变异位点。 如果这些序列中既有高度分歧也有非常相似的序列,MAXCHI偶尔会给这些密切相近序列提供假阴性结果。 在这些情况下,序列应该分成例如仅包含来自相同物种的序列的组,包含来自不同物种的一个代表性序列的组,包含来自不同属的一个代表性序列的组。(可以比对阶段或在RDP3中禁用某些序列)
5.CHIMAERA测试方法
CHIMAERA是David Posada对Maynard Smith最大χ2方法的修改。当沿着对齐序列的长度绘制时,这些χ**2值中的峰(figure12b)表示潜在的重组断裂点。与MAXCHI一样,CHIMAERA提供有关潜在断点的位置,但未提供有关重组区域的范围。**
CHIMAERA和MAXCHI之间的区别在于:(1)选择多态性位点的方式不同;(2)CHIMAERA只能用于三元组序列的重组筛选。
当检查差异大的序列时,CHIMAERA不会遇到与MAXCHI相同的问题,但由于它依赖于亲本和重组序列之间的匹配,当在比对中仅存在一个亲本序列时可能难以识别重组。
(1)方法步骤
① 重组体序列的所有单态位点被丢弃,转换成1和0的线性串,1表示重组体与其中一个亲本的匹配,0表示与另一个亲本的匹配。
②分成两个分区的窗口以其中心沿着1和0的字符串一次移动一个位置。
③在每个窗口位置处,计算中央分区两侧的2 ×χ2值,用它来表达中央分区两侧的1和0的比例差。
原理图如下: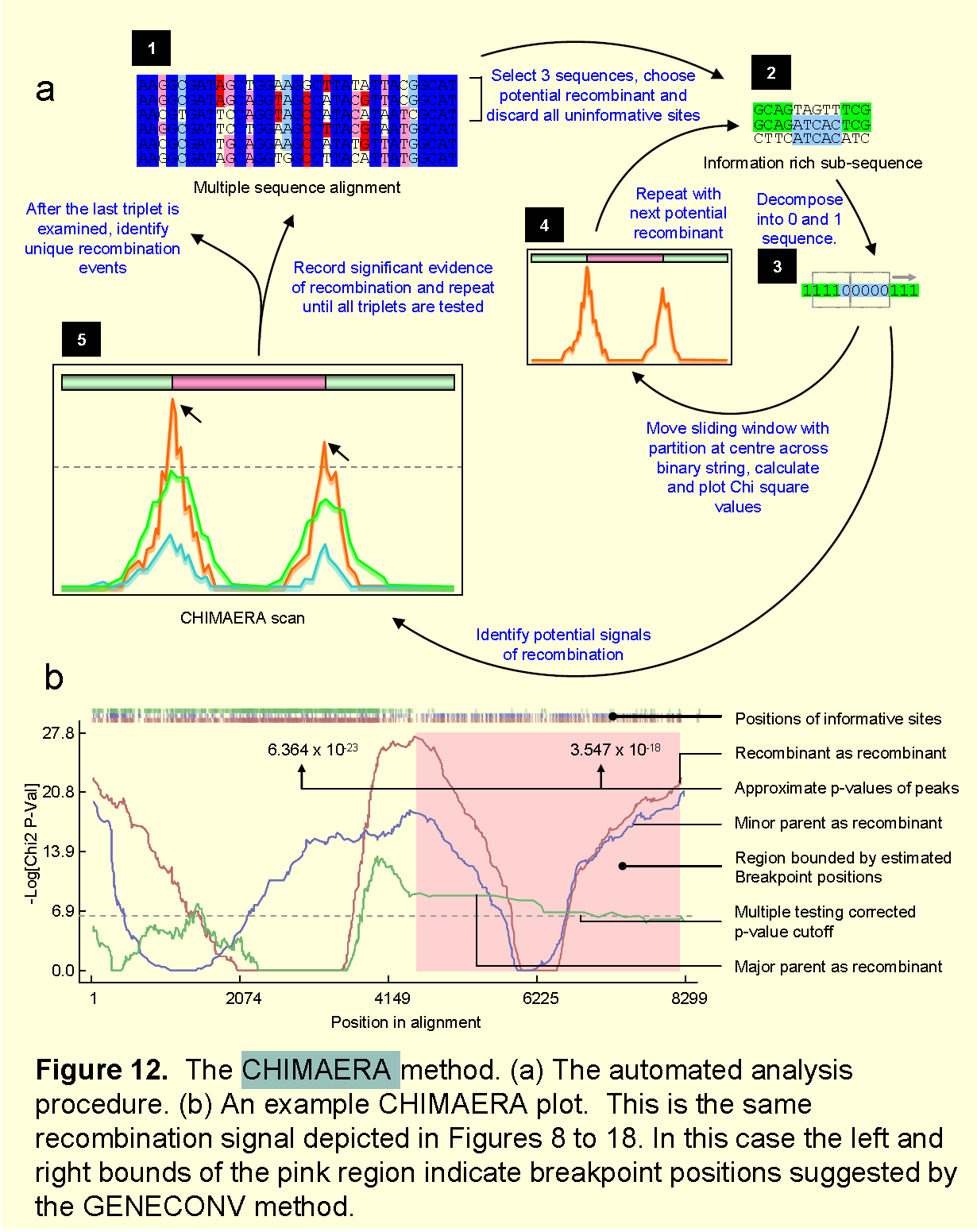
(2)注意
由于MAXCH和CHIMAERA之间的相似性,用这两中方法中的一种去确定另一种的检测结果可能不是一个好主意 ,言外之意是 通过MAXCHI和BOOTSCAN方法检测到的重组信号比通过MAXCHI和CHIMAERA方法检测到的重组信号更好。
6.SISCAN检测方法
(1)方法步骤
①第四条序列通过三元组序列中的某条序列“水平”随机化构建,或者从对齐的序列中选取(对齐的序列中最分歧与三元组序列最密切相关的序列)。但与三元组的三条序列相比,这些“水平”随机化构建彼此之间的距离更远 - 即最近的异常值。
②设定长度的窗口沿着四条序列一次移动一定数量的核苷酸。如果使用随机序列,则每个窗口会产生新的随机序列(由称为水平随机化的过程构建,其维持核苷酸含量)。
③把对齐序列的每列序列定义为十五个不同类别中的一个。
④以用户定义数量的置换比对将每列序列的核苷酸随机化(称为“垂直”随机化。针对每个置换对准确定落入十五个不同类别的每列序列。
⑤每个窗口,使用Z检验来确定该窗口中与15个位点类别中的任何一个对应的列数与垂直随机序列是否差异显著。
原理图如下: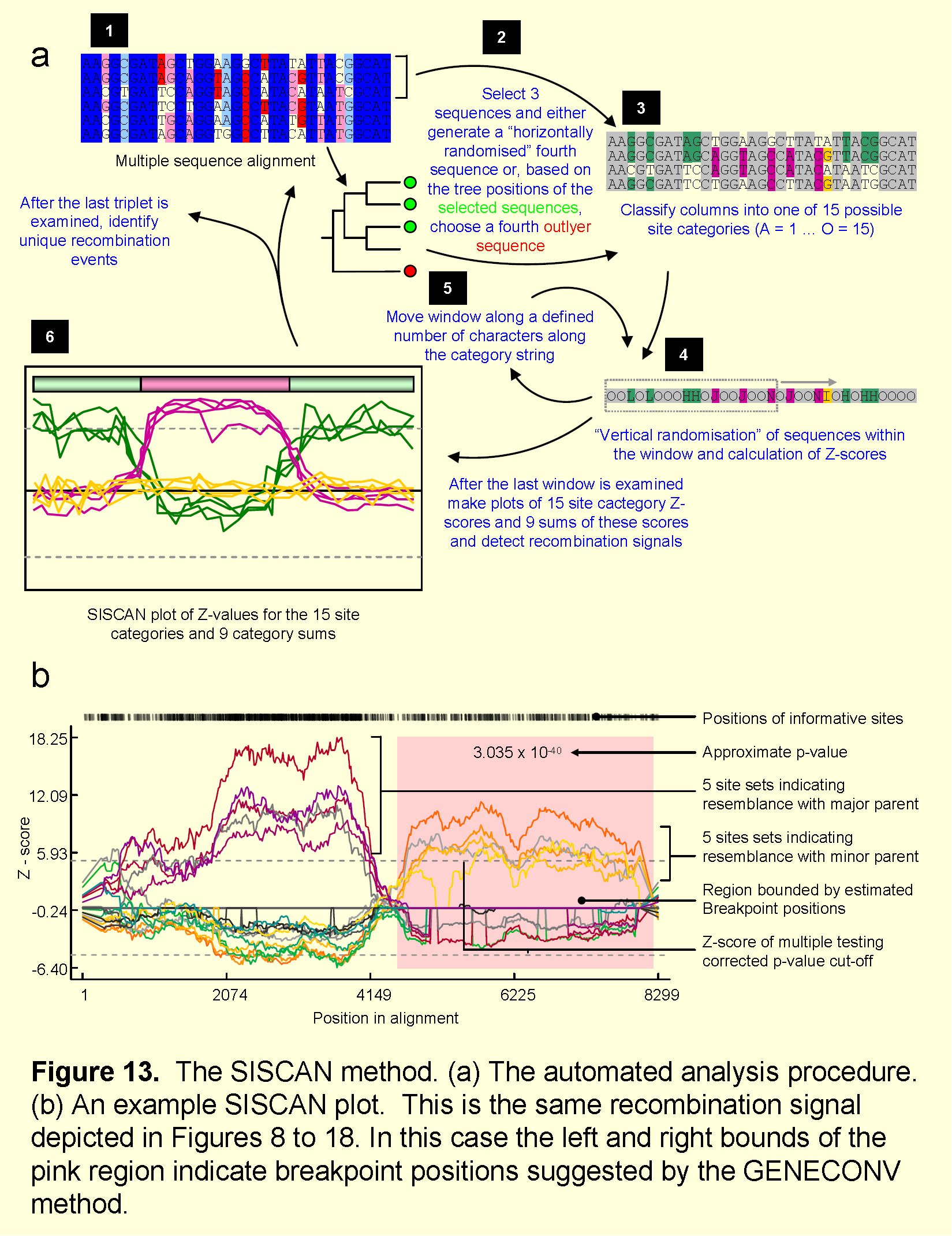
(2)潜在问题
SISCAN的主要问题是它像BOOTSCAN一样,不仅仅是变异位点,会检查所有位点。 虽然该方法可以设置为“ignore”不变位点,但在整个分析过程中,SISCAN窗口大小相对于底层对齐保持不变。 这意味着在对齐序列中的变化较小的部分(或者当检查发散较少序列时),每个窗口可能有太少的位点使分析无效。 增加窗口大小以适应较少的可变区域可以一部分解决该问题;但是对于较大的窗口,来自小的重组区域(小于窗口大小的一半)的重组信号将更难以检测,可以尝试设置窗口大小,使每个窗口平均包含10到20个可变站点。
7.3Seq检测方法
3SEQ是由Maciej Boni(Boni等人,2007)开发的三元组扫描方法(如BOOTSCAN / RECSCAN,RDP,MAXCHI,CHIMAERA和GENECOV)。有关3SEQ的更多信息,请参阅http://www.cggh.ox.ac.uk/3seq-source/3seq_manual.pdf上提供的3SEQ用户指南。
与MAXCHI,CHIMAERA,GENECONV和RDP方法一样,3SEQ仅关注从较大的对齐序列中得到的序列三元组的多态性位点。事实上,由3SEQ(figure14a)检查的位点与CHIMAERA检查的位点完全相同(figure12a)。然后使用以下步骤查询三元组中的每个序列以确定它是否可能是三元组中其他两个序列的重组体。软件说明书http://www.cggh.ox.ac.uk/3seq-source/3seq_manual.pdf.
(1)方法的步骤
①重组体的所有的单态位点及和亲本匹配的位点被标志成+ 1和-1,其中+1表示重组体与其中一个亲本的匹配,-1表示与另一个亲本的匹配。
②从-1和+1序列的每一端开始,在每个新位置记录-1和+ 1的运行总和。言外之意,持续增加表示和某一条亲本越来越近,持续减少也表示和某一条亲本越来越近。
③然后记录序列中任何两个位点的运行总计的最大差异以及位点之间的距离。
④运行中变化最大的位点边界可能重组的断点,运行中记录的差异和核苷酸分离的数量用来计算p值。
原理图如下: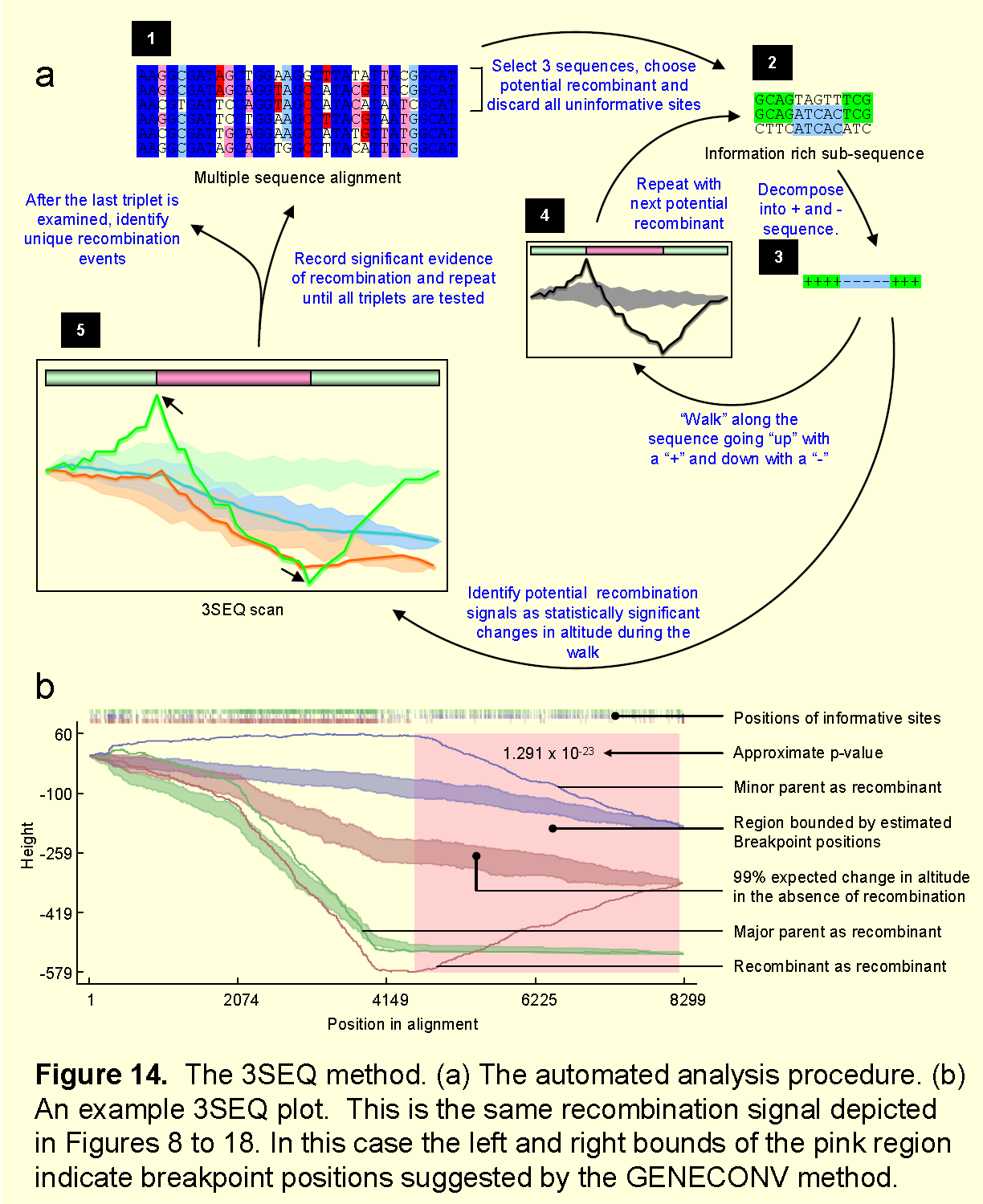
(2)该方法潜在问题
与CHIMAERA检测方法一样,3SEQ方法依赖于亲本和重组序列之间的匹配,并且当在比对中仅存在一个亲本序列时可能难以识别重组。<br /> 因为3SEQ和CHIMAERA在寻找重组时查询完全相同的核苷酸位点组合,所以用3SEQ和BOOTSCAN方法比只通过3SEQ和CHIMAERA方法检测能检测到更加能被证明的重组信号。
8.PHYLPRO检测方法
PHYLPRO方法是一种小区域重组的检测方法,还可用于测试其他方法识别断点位置的准确程度。但是因为它不能去量化重组信息,所以不能用于重组的自动探索性扫描。
(1)该方法检测步骤
①PHYLPRO与MAXCHI和CHIMAERA方法一样,在PHYLPRO设置里划分一定长度的窗口,在窗口区域的中心沿着对齐序列的一次移动一个核苷酸。
②对于每半个窗口,估算序列之间的Hamming or p-distance。
③左半窗口(或右半窗口)某条序列与其他序列距离的测量采用Pearson回归系数(R)。
④除了最低的回归系数R值可能与重组断点有关外,有着最低回归系数值的序列可能是重组序列。
原理图如下: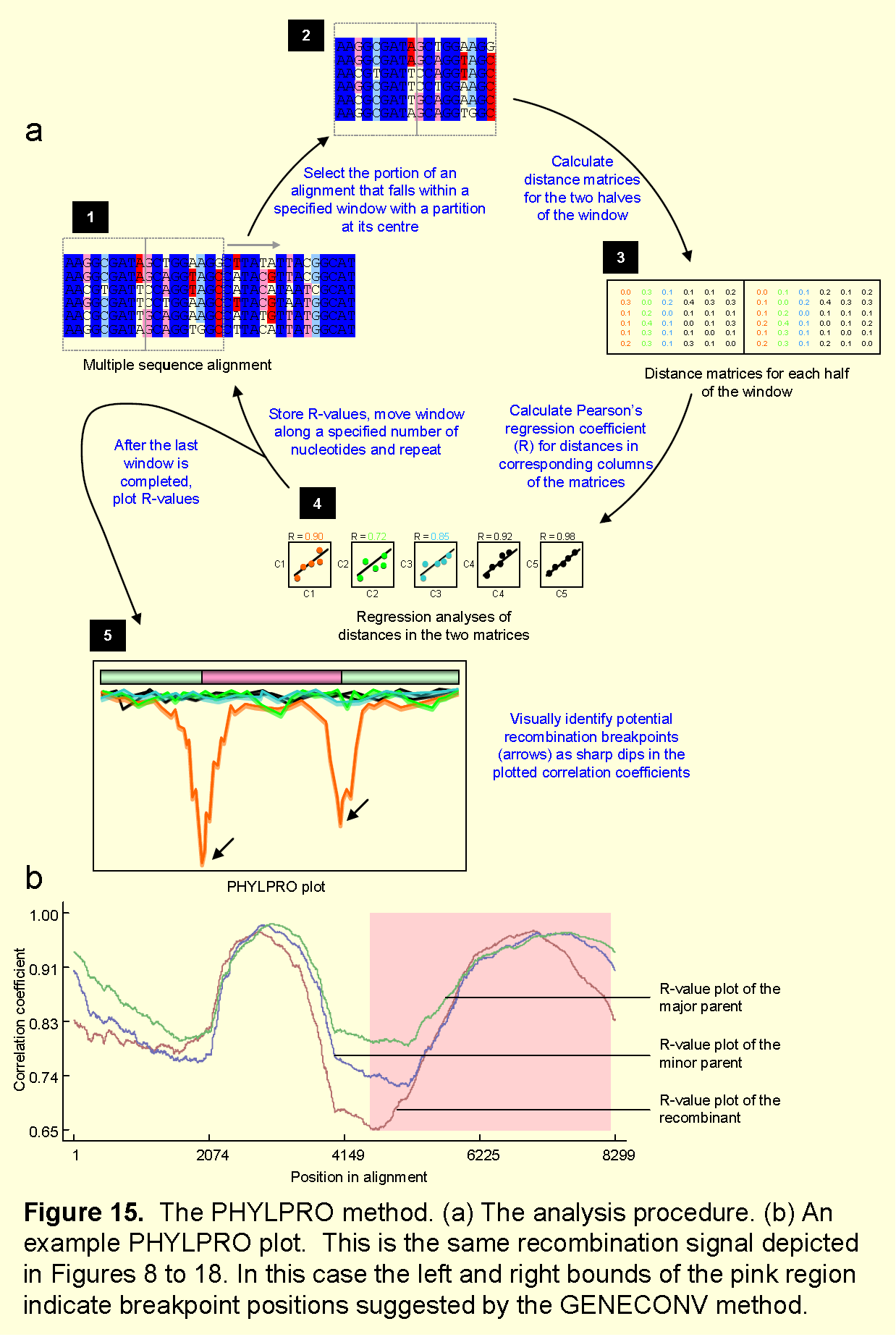
(2)潜在的问题
PHYLPRO方法主要的缺点是它不是一种能统计可能重组事件显著性的快速方法。当然,与Bootscan/RECSCAN 以及 SISCAN检测方法相比,PHYLPRO方法会查询所有序列位点而不只是多态性位点。RDP3软件作者没有确定PHYLPRO在确定断点位置方面是否比MAXCHI和CHIMAERA等相对准确的方法更准确。因此,对于识别重组断点最好使用MAXCHI和CHIMAERA方法。
9.VisRD检测方法
VISRD是一种直接鉴定小区域重组的检测方法(另外一种是PHYLPRO ),它会自动连接其他方法检测到的重组事件,还可以构建 “Highway”和“Occupancy”图,对其他方法检测到的重组信息再次检测。请注意,在RDP3中的VisRD的功能不完整,独立的VisRD3.0程序中有许多可用的功能,VisRD3.0程序下载链接:http://www.uea.ac.uk/cmp/research/cmpbio/Phyologenetics+Software+-+VisRD,使用手册:http://www.uea.ac.uk/polopoly_fs/1.122536!Visrd_manual.pdf.
(1)该方法检测的步骤
①从较大的对齐序列中画出四条序列(称为四元组),并且一个固定宽度的窗口沿着这些序列一次移动一个核苷酸;
②在每个窗口中,分别计算对这四条序列的三种可能的树拓扑结构support值, 每种树拓扑结构对应三角形上的一个角,每种树拓扑结构的support值映射为三角形上的点;每个位置与角之间的距离与其对应的拓扑结构support值成比例;
③重组断裂点的任一侧的四元组序列的三种拓扑结构映射到三角形中每个点坐标的改变,可以用于指示哪个序列是重组的。
比如:60bp长度的4条待检序列对应的a,b,c三种拓扑结构对应三角形的A,B,C三个角,每种拓扑结构的support值对应角周围的一个点,这样在A,B,C三个角处会得到3个点;然后窗口沿着这些序列一次移动一个核苷酸,比如现在这4条待检序列长度变成了61bp,这4条序列三种拓扑结构对应的support值又会在A,B,C三个角处产生新3个的点,如此窗口不断移动,每次不断在A,B,C三个角周围产生3个新的点,那么最后拥有点最多的那个角对应的拓扑结构即为这4条待检序列最可能的拓扑结构。因为每种拓扑结构里记录了亲缘关系(比如60bp-120bp的片段,a拓扑结构里序列1和序列2最近,如果2是重组体,那么1就是主要亲本),所以这些点的数量既可以反映每种拓扑出现频率,还可以反映亲缘关系最近的两条序列的出现频率。
即occupany值对应某种拓扑结构的出现频率,也对应亲缘关系最近两条序列的出现频率。举个例子,对于待测的4条序列,如果在60bp-120bp处occupany值忽然下降,则是60bp-120bp的拓扑结构与前一区域(0-60bp)的拓扑结构发生了很大变化,同时亲缘最近的2条序列也发生了变化,比如0-60bp是1序列和2序列亲缘最近,而61-120bp是2序列和3序列最近。这样就可以把重组断点,重组区域,主要亲本和次要亲本都找出来。
原理图如下: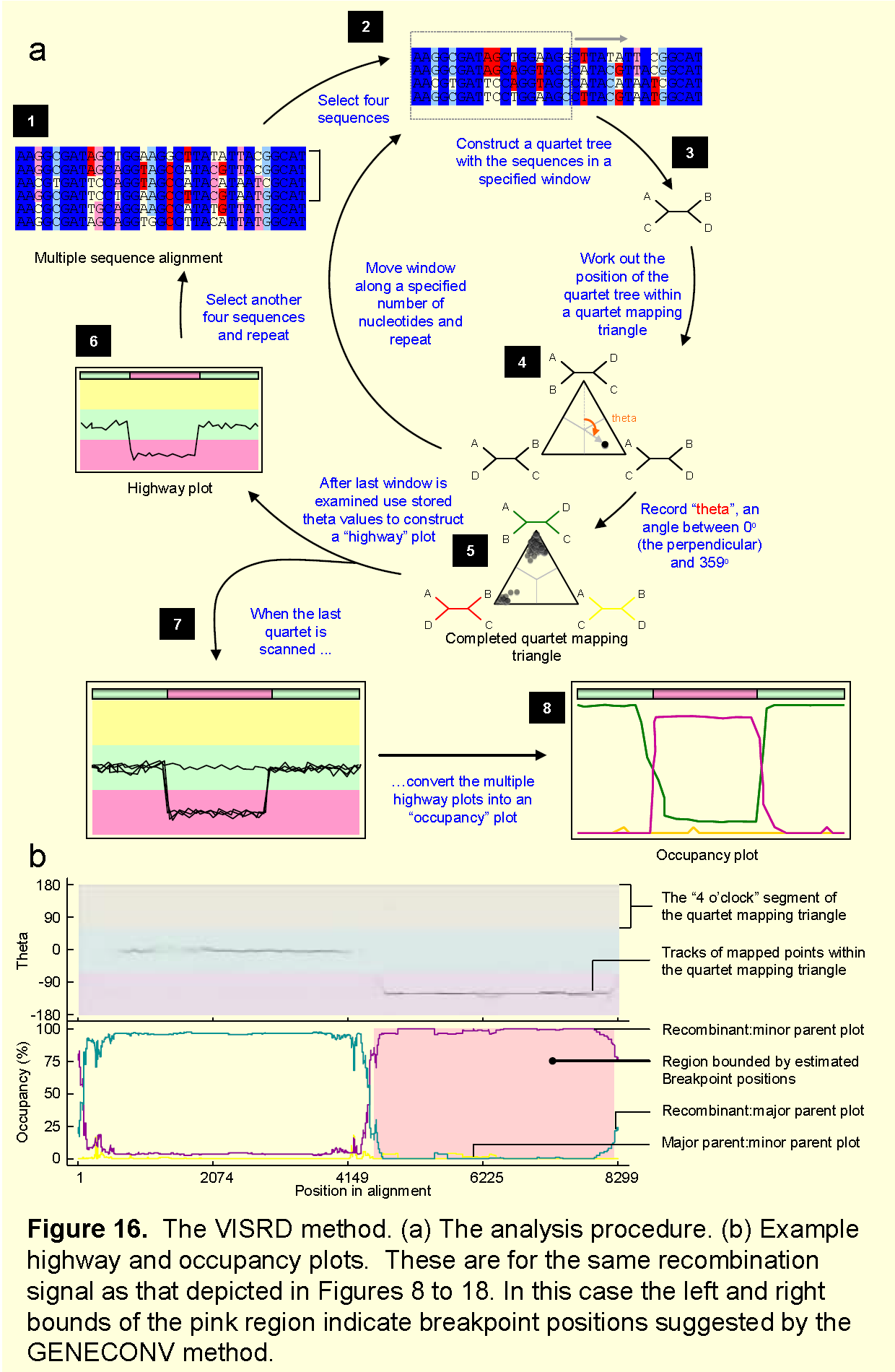
(2)潜在问题
VisRD没有简单的方法来测试潜在重组信号的统计显著性,虽然作者在更新的独立版本里解决了这个问题,但是在RDP3里不能测试潜在重组信号的统计显著性。
10.LARD检测方法
主要是识别重组断点,和MAXCHI方法有点相似。一次性描三条序列(重组和两个亲本序列),用于最佳地分离序列中具有不同的系统发育信号区域的点。
(1)该方法检测的步骤
①三条序列被分成两个分区(两个部分),通过最大似然法去建无根树,计算两侧分区建的树的枝长。
②通过似然比检验(评估6个参数的分区树和3和参数的未分区树)来对分区两侧的树的枝长进行变化(比如它们因为重组有不同的枝长),去获得高似然值;
③如上所述测试序列的每种可能的分区,在未分区树上似然值提高最大的点被认为是最可能的重组断点。
原理图如下: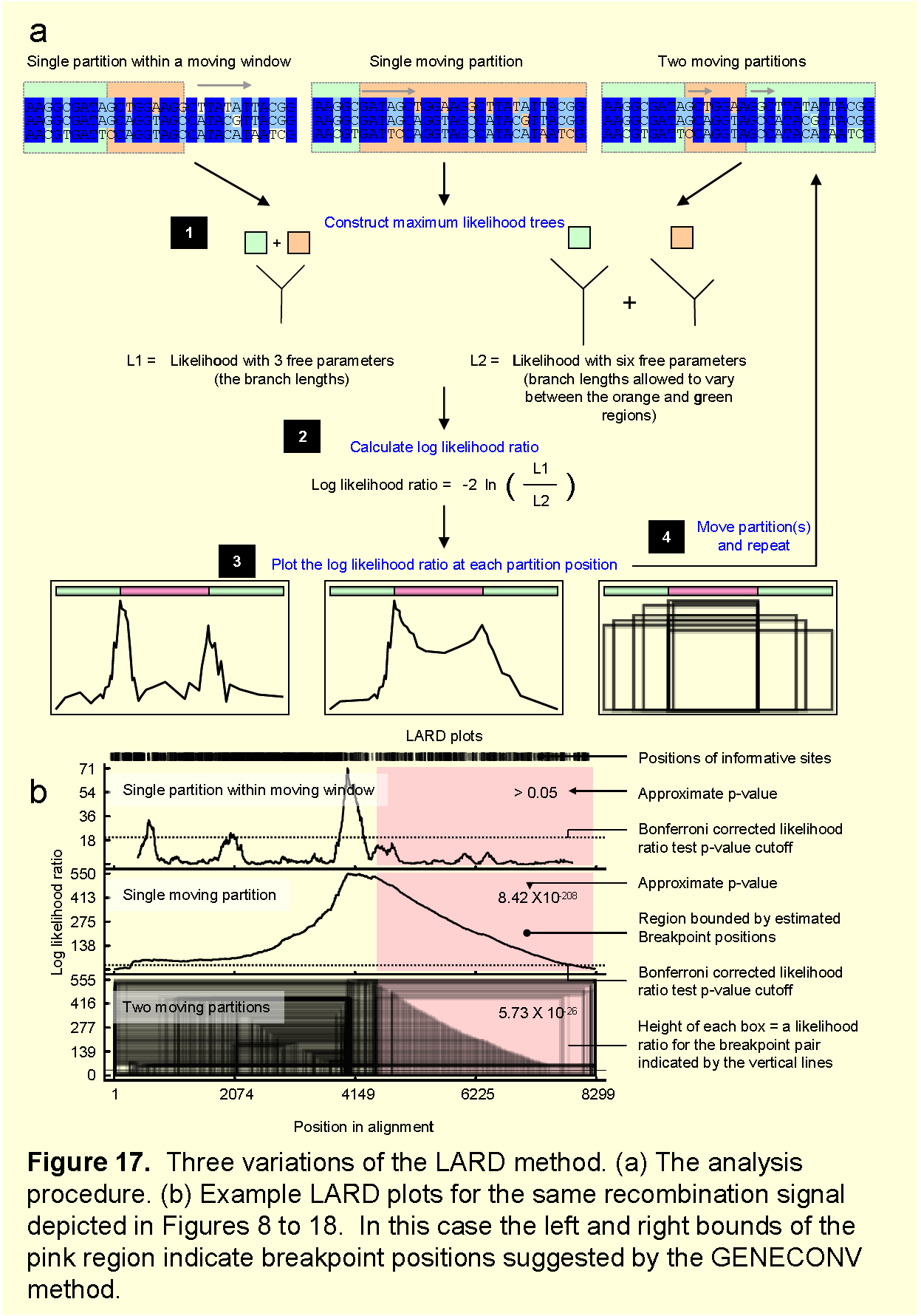
(2)存在问题
尽管LARD解释了位点之间的速率变异,但是它无法区分重组的序列与另外两个序列中的相应区域以更大或更低的速率进化的情况。
11.DNA Distance Plots
DNA距离图可用于提供潜在重组序列与其提出的亲本序列之间关系的图形描述。
(1)步骤
①所提出的亲本和重组序列按照设定长度的窗口沿着序列一次移动一定数量的核苷酸;
②每个窗口利用DNADIST (a component of the PHYLIP package) 计算Pair-wise distances,再相对于窗口中心对齐的位置去描点。
(2)注意
对于除“similarity”之外的所有距离模型,距离以“evolutionary units”测量,“evolutionary units”与序列之间发生的核苷酸取代的数量成比例但不等于。有人错误地采用了DNA距离图数据,从1中减去每个距离测量值,再乘以100,并将所得到的“percentage identities”图作为 “similarity plot”(由程序SimPlot绘制))。 有关DNADIST如何测量距离的更多信息,请参阅PHYLIP手册中的DNADIST文档:http://evolution.genetics.washington.edu/phylip/doc/dnadist.html
12.TOPAL/DSS检测方法
鉴于比对后的序列,TOPAL试图通过寻找相邻序列区构建的系统发生树的差异来鉴定重组断裂点。 它在某种程度上是LARD,BOOTSCAN和MAXCHI方法的混合体。
(1)步骤
①在具有分区窗口的中心,按照设定的长度滑动窗口,一次沿着序列移动一定数量的核苷酸。
②在每个窗口位置处计算距离矩阵(归一化到整个对齐序列的距离矩阵),并且为分区的任一侧上的序列构建NJ树或者或最小二乘树。最佳分支长度由未加权最小二乘确定,并且记录分区两侧的相应平方和和树拓扑的总和。
③交换每一个分区的拓扑结构,forced拓扑结构的最佳枝长通过最小二乘法计算,并记录平方和;
④针对每个分区记录forced和unforced树拓扑的平方和(DSS)之间的差异,记录每个窗口的DSS得分中的较高者。
⑤沿着序列长度的DSS峰指示潜在的重组断裂点。
⑥DSS峰值的显著性可以通过参数bootstrapping 来确定。
原理图如下: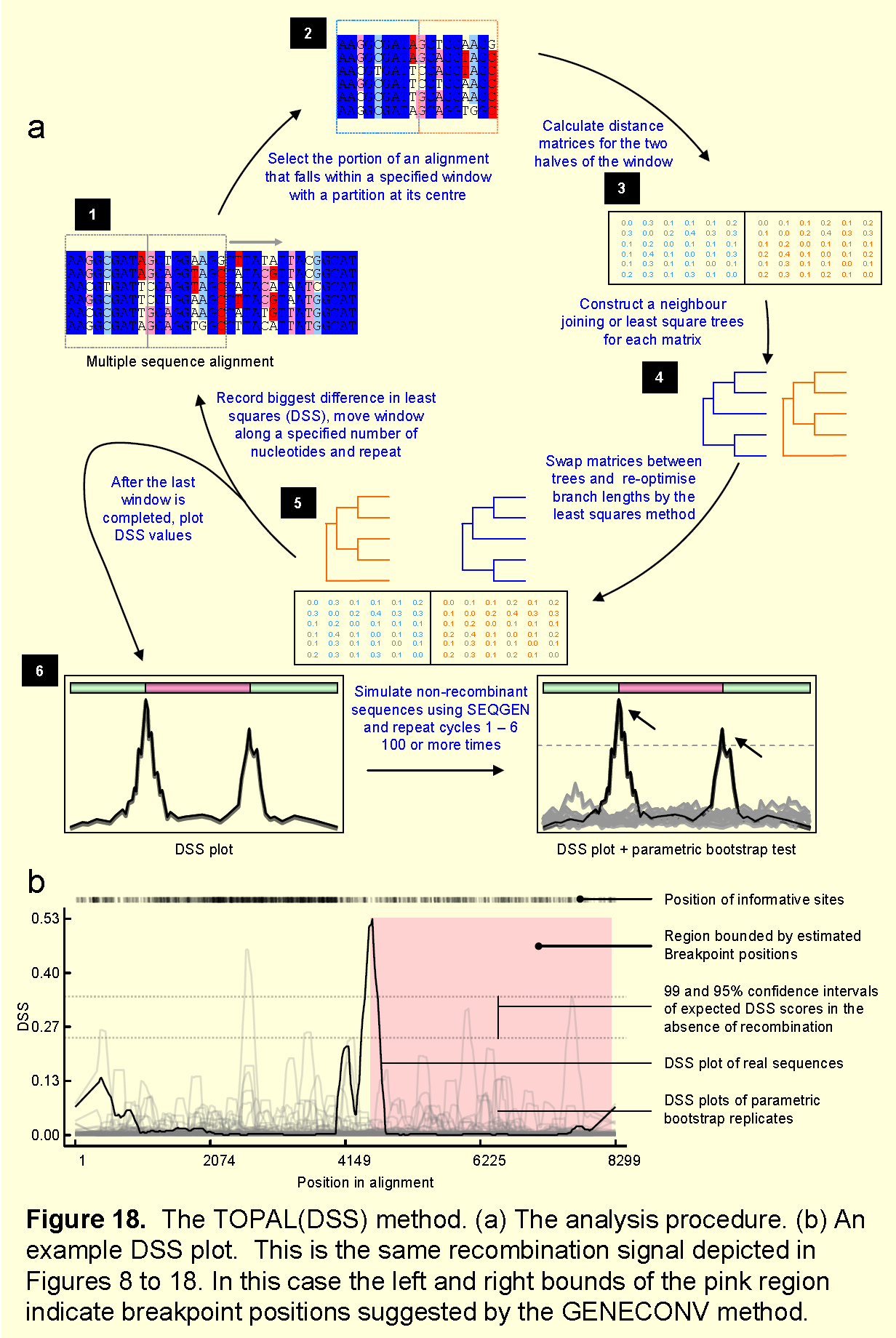
有关TOPAL算法的更多信息,请参阅McGuire和Wright(1998,2000)或TOPAL手册,该手册可在线获取:http://202.38.126.65/mirror/www.bioss.sari.ac.uk/~frank/Genetics/welcome.html
RDP3的TOPAL方法可用于再次检查其他方法检测到的重组结果(原始RDP,MAXCHI,GENECONV,BOOTSCAN,CHIMAERA,SISCAN和3SEQ方法),TOPAL中每一次只能检查三条序列。当使用仅包含三个序列的树时,平方和计算(由PHYLIP包的FITCH组件执行)通常不会产生任何结果(例如,当三个分支中的两个具有相同的长度时)。
为了“解决”这个问题,RDP3生成了第四条序列,它是由原始对齐序列随机化而来,(用于生成序列的随机数与在TOPAL分析的其余部分期间使用的随机数相同),通过沿着序列一次移动一个核苷酸,并从该位置处的序列中随机选择核苷酸来产生第四个序列。
TOPAL不能用于重组的自动分析的原因是该方法的参数自举部分(需要推断DSS峰值是否代表重组断点的显著性)非常慢。如果对使用TOPAL进行自动检测重组感兴趣,建议尝试将其从“checking”方法升级为“automated screening”方法。
所以,TOPAL是在其他方法鉴定为可能重组后,再在结果面板的左下角部分,手动选择TOPA分析方法去分析。
(2)潜在问题
产生第4条序列的一个问题是:该序列实际上是所有对齐序列中其他序列的随机重组体。它取决于所有序列的数量及其彼此的相关性,第四序列可能不是所有对齐序列的合适“平均值”;例如,如果序列中包含大量彼此密切相关的序列和少量距离较远的序列,那么这条随机序列将更加相似于密切相关序列组中的序列;因此使用TOPAL检查距离较远的序列之间的重组时,这可能存在问题。但请注意,此问题特定于TOPAL的“check using”版本,并且“Manual TOPAL scan”版本不会出现问题,该版本应与原始方法的工作方式相同。
四. 检测方法特点速查表
| 检测方法 | 扫描特点 | 主要功能 | window sizes的含义 | 扫描的窗口是否分区 | 备注 | step size |
|---|---|---|---|---|---|---|
| RDP | 一次扫描3条序列的仅变异位点 | 识别重组区域。 A-C或B-C的indentity百分比高于A-B的区域被鉴定为潜在的重组区域。 | 每个扫描窗口包含的变异位点数量,默认值是30 | 否 | G-scale对错配的核苷酸评分。 | 无 |
| GENECONV | 一次扫描3条序列的仅变异位点 | 识别重组区域。高P值对应高匹配得分区域。 | 无 | 否 | 无 | |
| BOOTSCAN/RECSCAN | 扫描序列的变异位点和保守位点 | 识别重组区域和重组断点。在两条不同序列之间交替的高bootstrap值(figure10b)的地方指示可能有潜在的重组。 | 每个扫描窗口包含的核苷酸数量,默认值是200 | 否 | 检测具有20个variable核苷酸位点的重组区域的最佳窗口大小是40。 | 推荐小于window sizes值的50% |
| MAXCHI | 一次扫描3条序列的仅变异位点 | 识别重组断点,但未提供有关重组区域范围的信息。沿着序列的长度绘制时,这些χ2值中的峰表示潜在的重组断裂点。 | 每个扫描窗口包含的变异位点数量,默认值是70 | 对称分成2个 | 如果三条序列的总变异位点数少于窗口设定值的1.5倍,则窗口设定值将自动调为总变异位点数的0.75倍。如果设定值小于10,则不扫描检测。 | 无 |
| CHIMAERA | 一次扫描3条序列的仅变异位点 | 识别重组断点,但未提供有关重组区域范围的信息。沿着序列的长度绘制时,这些χ2值中的峰表示潜在的重组断裂点。 | 每个扫描窗口包含的变异位点数量,默认值是60 | 对称分成2个 | 如果三条序列的总变异位点数少于窗口设定值的1.5倍,则窗口设定值将自动调为总变异位点数的0.75倍。如果设定值小于10,则不扫描检测。 | 无 |
| SISCAN | 一次扫描4条序列变异位点和保守位点 | ? | 每个扫描窗口包含的核苷酸数量,默认值是300 | 否 | 要求小于window sizes值的50% | |
| 3Seq | 一次扫描3条序列的仅变异位点 | 识别重组断点,运行中变化最大的位点边界可能重组的断点。 | 否 | |||
LARD |
一次扫描3条序列变异位点和保守位点 | 主要是识别重组断点,和MAXCHI方法有点相似,在未分区树上似然值提高最大的点被认为是最可能的重组断点。 | 每个扫描窗口包含的核苷酸数量,默认值是400 | 非对称分成2个 | 窗口大小设置应足够大,每个检查的窗口包含至少20个变异核苷酸位点,因为窗口被分成两部分,并且其最佳的窗口大小是BOOTSCAN和SISCAN方法的两倍。 | 设置为10应该是一个很好的折衷方案 |
| PHYLPRO | 扫描序列的变异位点和保守位点 | 识别重组断点,最低的回归系数R值可能与重组断点有关,有着最低回归系数值的序列可能是重组序列。 | 每个扫描窗口包含的变异位点数量,默认值是60 | 对称分成2个 | 窗口大小设置应足够大,每个检查的窗口包含至少20个变异核苷酸位点。 | 无 |
| DNA Distance Plot | 一次扫描3条序列变异位点和保守位点 | ? | 每个扫描窗口包含的核苷酸数量,默认值是200 | ? | 根据用于检测的亲本的相关性来设置窗口大小。每个窗口应包含至少5个可变位点。 | 最佳值取决于被检查序列的相关性,并且应小于window sizes的20% |
| DSS (TOPAL) |
扫描序列的变异位点和保守位点 | 主要是识别重组断点,沿着序列长度的DSS峰指示潜在的重组断裂点。它在某种程度上是LARD,BOOTSCAN和MAXCHI方法的混合体。 | 每个扫描窗口包含的核苷酸数量,默认值是200 | 对称分成2个 |
窗口大小设置应足够大,每个检查的窗口包含至少20个变异核苷酸位点,因为窗口被分成两部分,并且其最佳的窗口大小是BOOTSCAN和SISCAN方法的两倍。 | 默认值10 |
| VisRD | 扫描序列的变异位点和保守位点 | 识别重组区域, occupany值对应某种拓扑结构的出现频率,也对应亲缘关系最近两条序列的出现频率。 | 每个扫描窗口包含的核苷酸数量,默认值是400 | 否 | 窗口应包含10个或更多变异位点。 | 无 |
Breakpoint Distribution Plot |
? |
? |
默认值是200 |
? |
小窗口尺寸(<= 50nts)对于检测异常紧密的断点簇(即高度聚焦的重组热点)是有用的,但对于检测重组冷点或分散的重组热点不是很好。窗口大小在100到200 nts之间通常是检测热点和冷点之间的良好折衷,但可能会错过小于指定窗口大小的区域内异常紧密的断点簇。 | 无 |
| Matrix | ? | ? | 指圆的直径,即断点对矩阵上每个重组断点对绘制的圆。默认值是200 | ? | 无 |
By zzj<br /> 2018年11月26日初稿。<br />转载请注明出处。<br />**如有问题可及时与我反馈,一起交流学习。**

若有收获,就点个赞吧
0 人点赞
