1.MEGA(找核苷酸模型或氨基酸模型)里的模型基于BIC标准,BIC值越低对应的模型越好。文件要求:fasta或者.mas格式的文件。
2.Mrmodeltest(找核苷酸模型)里有两个标准,一个是hLRTs,另外一个是AIC标准,优先选择AIC标准下的最佳模型。(软件结果会直接给出使用该模型建树的代码,可以直接复制到文件里用于PAUP或者Mrbayes建树),文件格式要求:连续的nexus文件。
3.Modeltest(找核苷酸模型),同Mrmodeltest。
注意:Modeltest和Mrmodeltest的运行依赖于软件PAUP,这一点非常不方便。
4.Jmodeltest(找核苷酸模型)里是采取计算似然值来找模型,用户可以自定义根据AIC或者BIC或者DT去找找最佳模型,一般选用AICc(AIC二级修正)或者BIC标准去找最佳模型。文件格式要求:.fasta格式或者连续的nexus文件或者phylip文件。(软件对应的文献给出了图形使用说明书)
5.ModelGenerator(找核苷酸模型或氨基酸模型)里优先选择AIC1下的最佳模型。文件格式要求:.fasta文件。
6.Prottest(找氨基酸模型)是通过计算似然值来选模型,它提供了不同的算法去计算似然值,得到结果后,以AICc=0的模型为最佳模型。文件格式要求:推荐使用.fasta格式或者连续的nexus文件。
7.Modelfinder(找核苷酸或氨基酸模型),常与建树软件IQ-Tree连用。Modelfinder是2014年新出的软件,很快,推荐使用。
8.关于序列文件的格式的转换,推荐软件BioAider里的Seqformat Convertor功能(https://github.com/ZhijianZhou01/BioAider)。切记,所谓的.nex格式也好,.fasta格式也好,.phy格式也好,指的是里面内容的布局,而非是这个文件的名称后缀。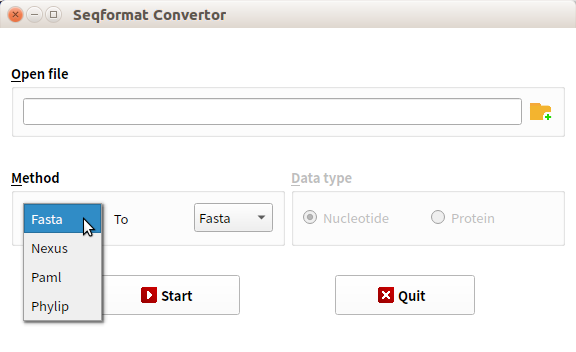
测核苷酸序列模型软件推荐:Modelfinder。
测氨基酸序列模型软件推荐:Modelfinder。

若有收获,就点个赞吧
0 人点赞
