前言: Beast是用来进行贝叶斯MCMC分析的一系列合集软件,其主要组成程序有BEAUti、BEAST、Tracer、TreeAnnotator、FigTree,这几个软件的组合使用常用来重建MCC树(Maximum Clade Credibility Tree),即最大可信进化分枝树。此外,还有诸如SpreaD3、Tempest、PiBUSS、LogCombiner、TreeStat之类的分析软件。
本文分两个部分,暂时先依据软件说明书简单介绍Beast v1.8程序包里(BEAUti、BEAST、Tracer、TreeAnnotator、FigTree)各软件的功能及其参数的含义,再以示例数据重建带有分子钟的MCC树。
Beast功能较多,Beast其他软件包的使用及Beast的其他功能后期再整理上传,本文则主要介绍构建带有分子钟的MCC树。
参考文献:《A Rough Guide to BEAST 1.4》、《Bayesian hylogenetics with BEAUti and the BEAST 1.7》、《Relaxed Phylogenetics and Dating with Con dence》
一. 软件参数介绍
1. BEAUti
BEAST系列软件的特点总的来说是参数设置(BEAUti)、数据运行(BEAST)、收敛诊断(Tracer)、树的获取(TreeAnnotator)、树的展示和美化(FigTree)分别在不同的程序中进行,**而BEAUti便是这众多步骤中的第一步,也是最为关键的一步。**
(1)打开BEAUti
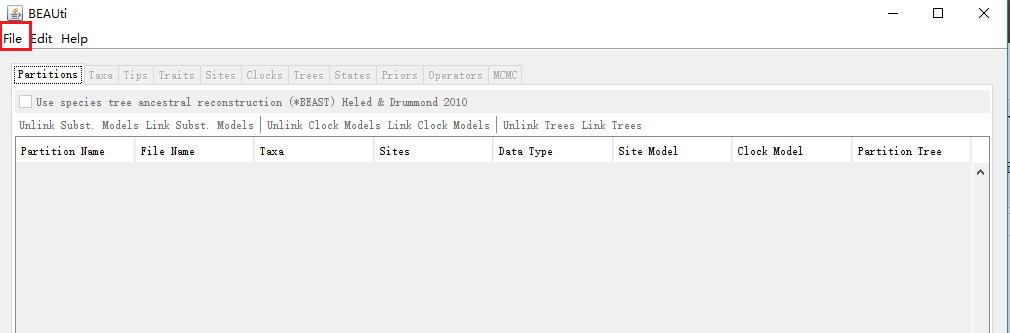 如上图,打开BEAUti后,通过点击“File”,选择“Import Data”导入待分析的序列数据,请注意必须是.nex格式的文件(该格式具体请参考下面附件)。
如上图,打开BEAUti后,通过点击“File”,选择“Import Data”导入待分析的序列数据,请注意必须是.nex格式的文件(该格式具体请参考下面附件)。
BEAST软件计算分化时间.pdf
如何将其他格式比如.fasta格式的序列文件转成.nex格式的序列文件?推荐使用软件BioAider里的Seqformat Convertor功能(https://github.com/ZhijianZhou01/BioAider)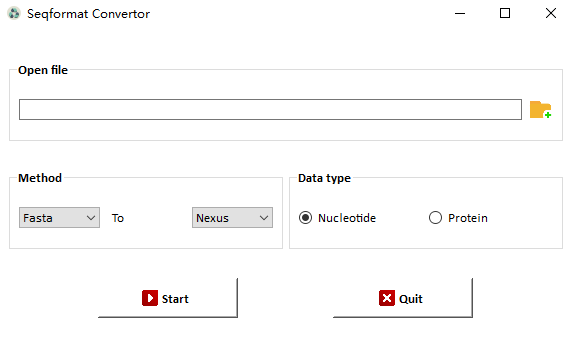
(2)导入.nex格式的序列文件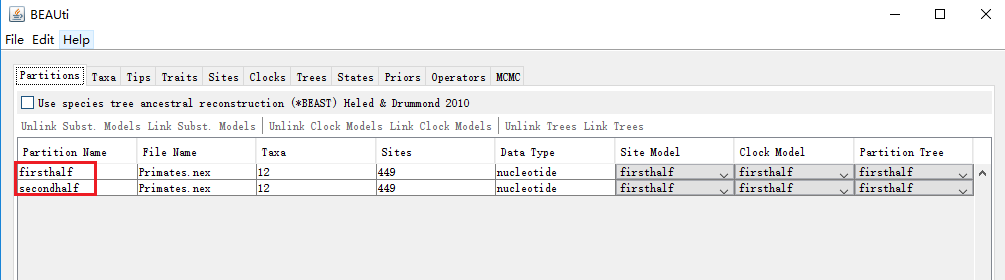
可以看到,上图红色方框部分,序列数据被分成了两个部分,这是因为在原始的.nex序列数据里对数据定义了分区,将序列数据分成了两个部分,如下: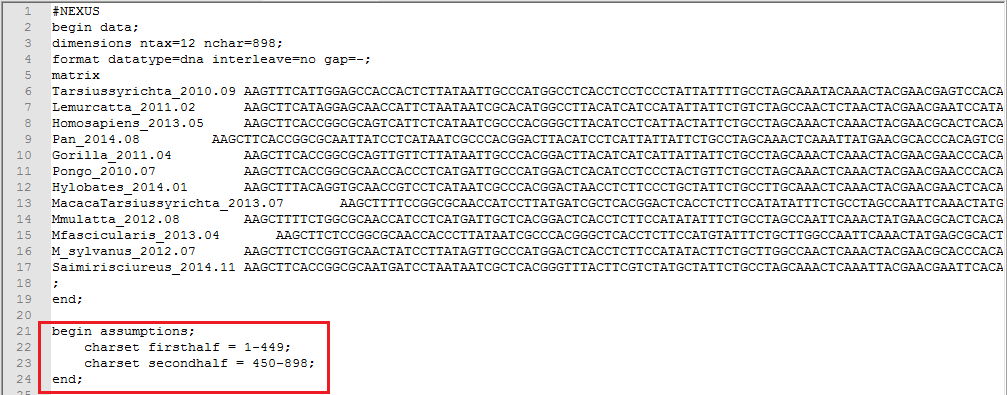 注:BEAUti也可直接导入fasta格式的序列,直接拖进BEAUti面板即可。
注:BEAUti也可直接导入fasta格式的序列,直接拖进BEAUti面板即可。
(3)Taxa选项(节点校准)
这个选项用可以在序列数据中设置分类单元子集,并**估计**设置的**分类子集**的**最近共同祖先分化时间(time to most recent common ancestor,即tMRCA),**并且还可以在相应的分化时间上设置先验分布。这些分类子集可以代表多物种分析中的不同物种或者同一物种内在地理上孤立的种群。**请注意,设置为同一分类子集的序列,并不能保证该组会在最终的MCMC分析中,对于其他分类群是单系的。**<br />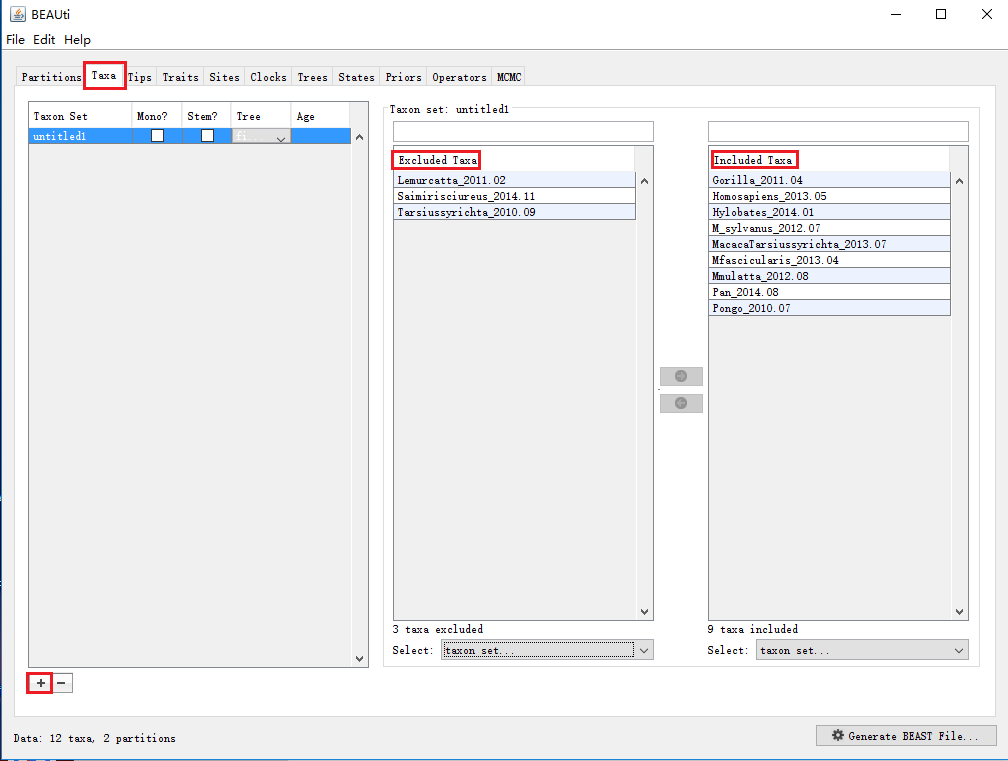“+”按钮添加分类子群,详解见上面附件“BEAST软件计算分化时间.pdf”,需要注意的是:**“Included Taxa”里的序列将用于估算tMRCA的日期。同时,还需要注意的是“Excluded Taxa”里的序列可能在也可能不在同一进化分枝里。**
Taxa选项的实际意义是:
(1)如果需要估算一些分类子集(谱系或者分枝)的最近共同祖先分化时间,可以在这里来设置,把需要估计的分类子集放进“Included Taxa”里;
(2)如果已知一些分类子集(谱系或者分枝)的共同祖先分化时间,可以用这些已知的信息来校正分子钟(又叫节点校准),也是同样选取分类子集,把它们放进“Included Taxa”里,之后在“Priors”菜单里设置对应tmrac参数的先验(范围)。
需要注意的的是,Mono(单系群)和Stem的校准点是不一样的: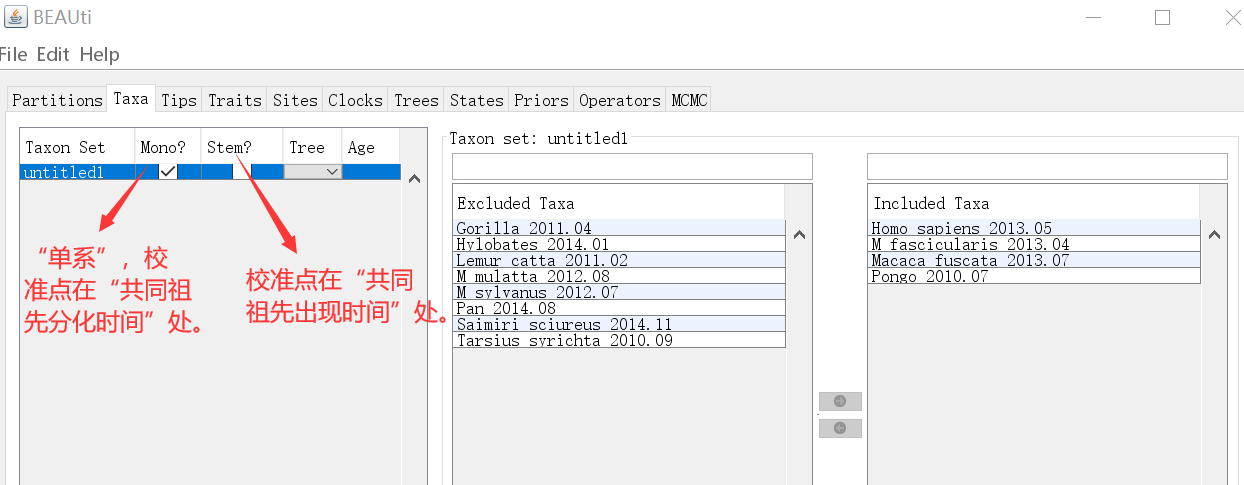
在进化树上体现如下图: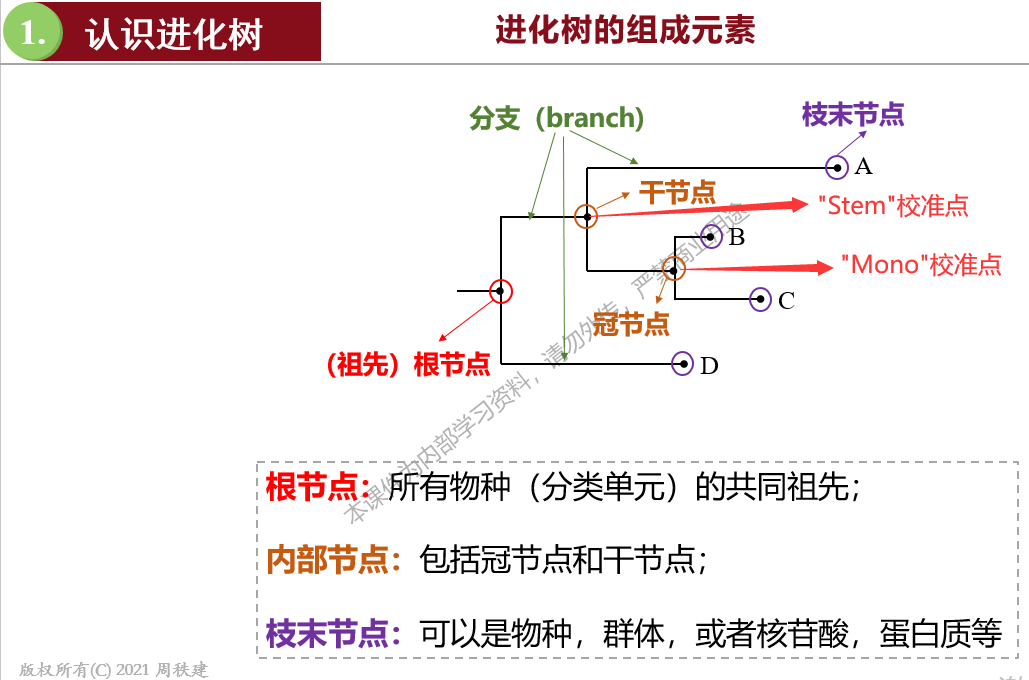
(4)Tip选项(端点校准)
该选项用来导入序列的采集时间,后期可以将序列采样时间用于分子钟校准。比如病毒这样的生物,我们可以使用它们的采样时间,填入beast里,来校准分子钟。
①点击“Tip”选项,在“Use tip dates”前打钩出现如下图: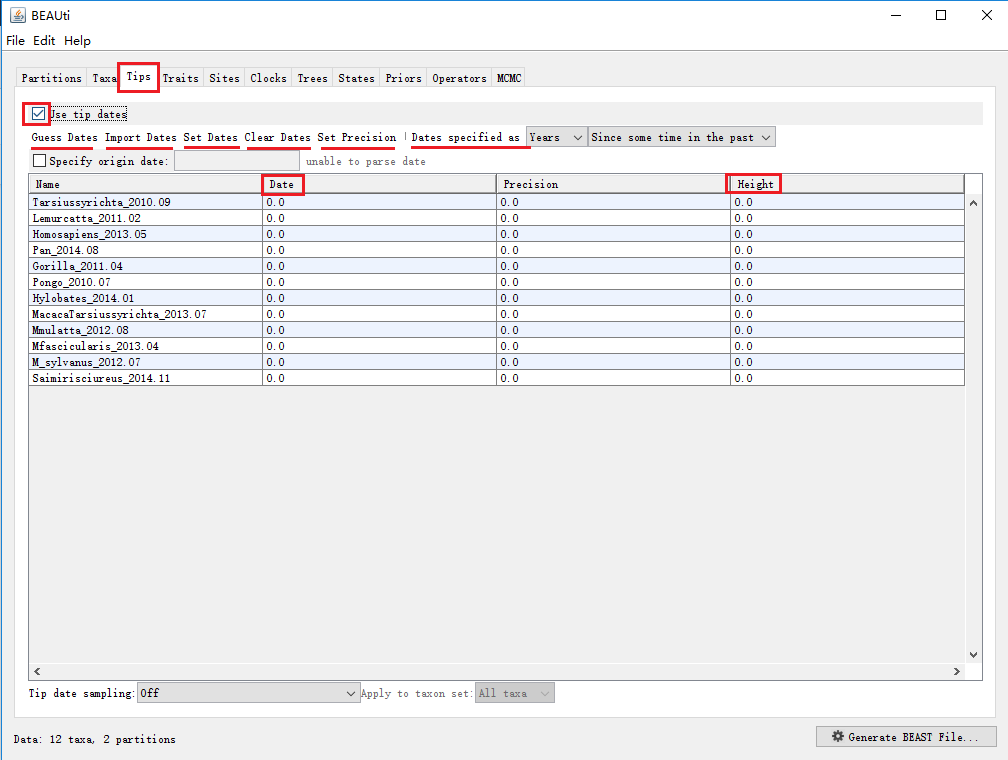 ②序列(名称)和采样日期的对应,可以通过导入单独的“序列名称和日期对应表”,或者对序列名称进行特殊的命名,再从序列名称里提前。本文示例采用后面一种方式:
②序列(名称)和采样日期的对应,可以通过导入单独的“序列名称和日期对应表”,或者对序列名称进行特殊的命名,再从序列名称里提前。本文示例采用后面一种方式:
如果你在序列名称里加了时间信息,可以自动获取,比如你的序列名称为Tarsiussyrichta_2011.09这样的年份.月份的格式,可以使用“Guess Dates”选项将第1个“_”后面的字符读取为采集时间(日期),同时将日期格式改为yyyy.MM。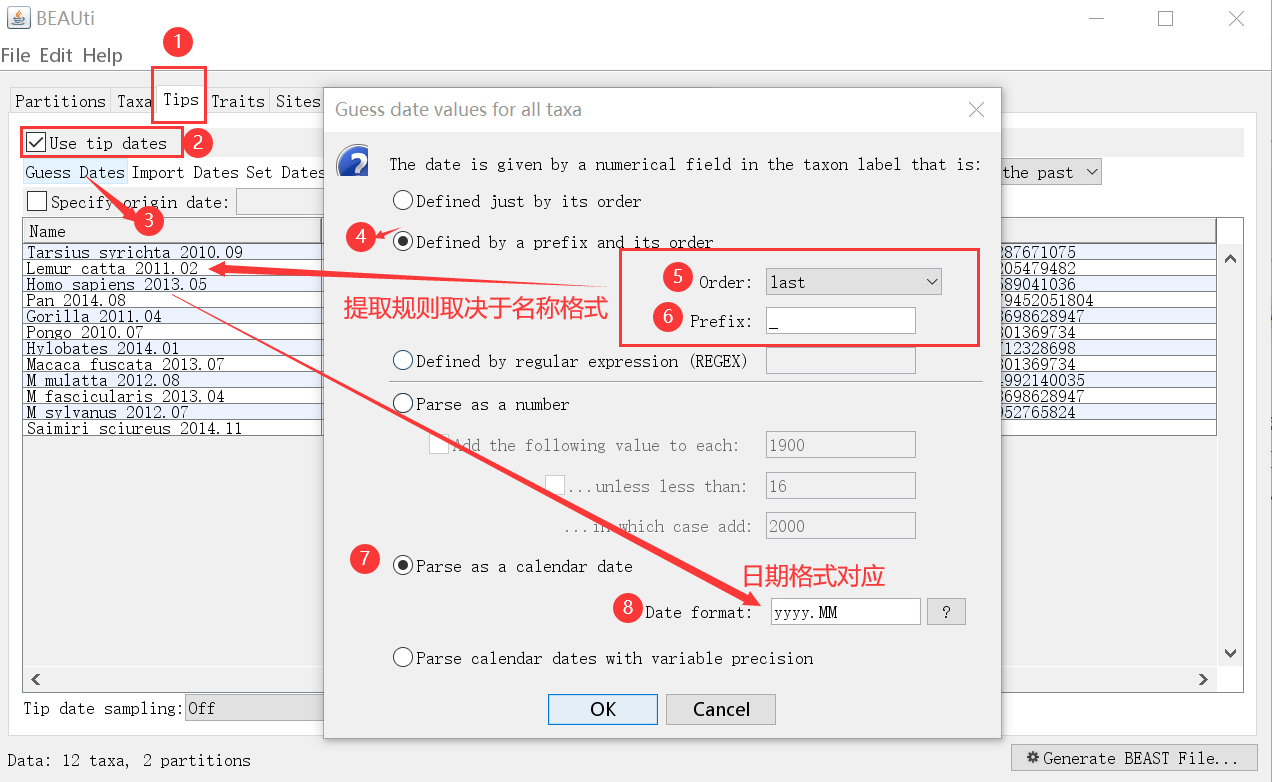
采样日期识别如下: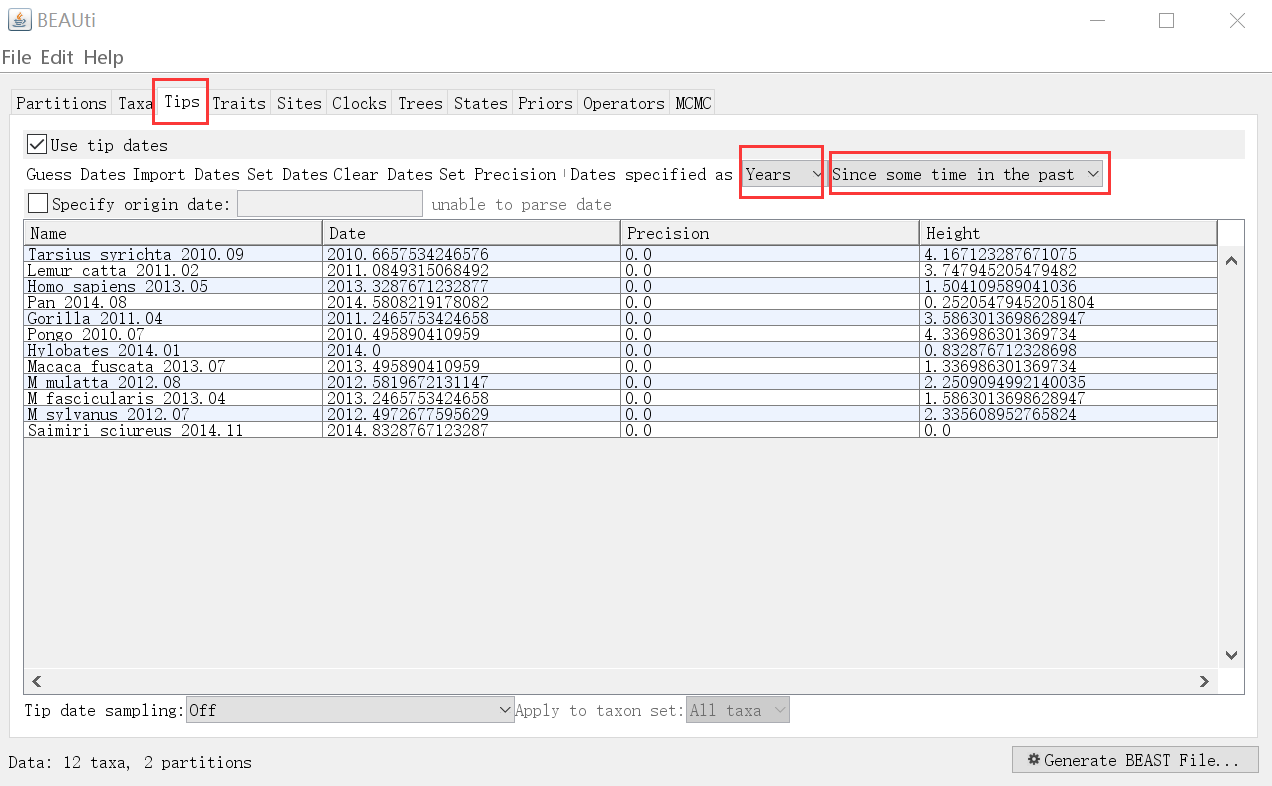
此时“Date”这一列显示的是日期的十进制格式,这里要注意稍微检查下日期转换是否有明显的错误。
注:因为示例数据是病毒数据,且是以“年”为单位,因此这里要选择“Years”和“Since some time in the past”。
For radiocarbon dates(, only enter the absolute date value, not the associated error. The dates entered need to be specified as “Years” and “Before the present” from the “Dates specified as” menu.
For viral data sets, dates will most commonly be calendar years (e.g., 1984, 1989, 2007, etc.) or perhaps
months or even days since the start of the study and need to be specified as“Years”, “Months” or “Days” and “Since some time in the past” .
② 还可以为每个日期时间设置精确度,也就是误差范围,通过点击“Set Precision”来弹出设置。
以上图示例序列数据为例,因为其时间单位是“年”,所以如果设置为1,精确度则为1年。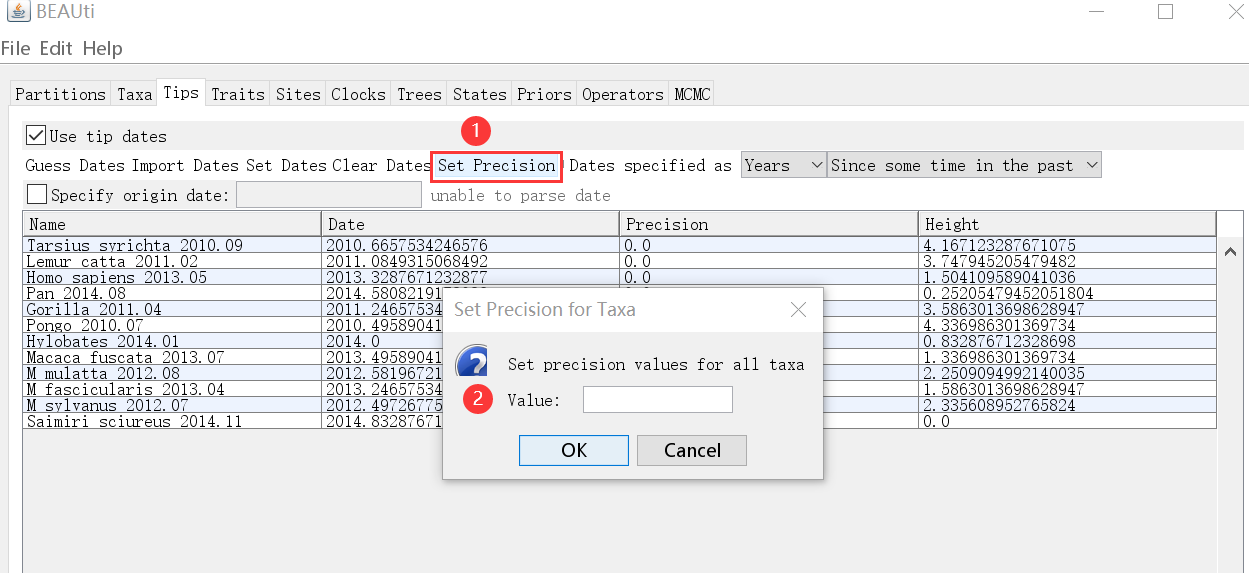
③ Height这一列指的是每个序列相对于最年轻序列的年龄高度。对于有时间数据的序列,软件会自动将最近采集的(即最年轻的,最现代的)序列的height(age)作为0(For non-dated sequences, all heights will be zero)。
④ 软件对数值单位保持全过程统一性
示例数据的日期是以“年”为单位,其实,不管使用什么时间单位来指定日期,beast都将在整个分析过程中保持一致。例如,如果你输入的是以百万年为单位的日期数值,这个时候0.1就代表实际的100,000年,那么所有结果里的日期和速率都将以 My(百万年)作为单位来提供。(例如,在My作为基本单位时,进化速率的数值0.01表示0.01 subs/site/My,换算成年份作为单位就是1.0E-8 subs/site/year)。
⑤ 对于日期数据处理的注意点:
(i)删除没有日期的序列;
(ii)找到有同位素标记的祖先DNA序列或者找有公布日期的DNA序列;
(iii)可以利用地理知识估算DNA序列的平均年龄;比如大多数沿海沼泽地矿床的年龄不到6000年,因此在这里发现的化石生物的平均年龄大约为3000年。
(5)进化模型设置
①对于分区数据的模型设置,因为不同分区可能设置不同的模型参数,故在设置模型前,先点击菜单栏“Partitions”,然后再Ctrl+鼠标左键选中两个分区,再点击“Unlink susets.Models”,即分区数据不关联模型。(非分区数据跳过该步骤)。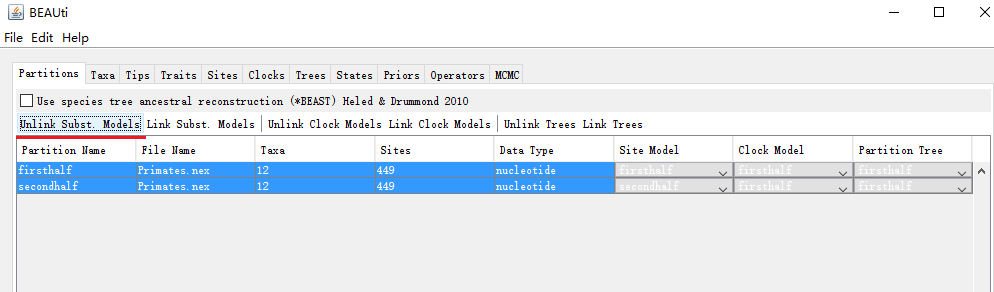 ②再点击菜单栏“Sites”设置进化模型
②再点击菜单栏“Sites”设置进化模型
进化模型主要有两部分组成,一是核苷酸(或氨基酸)替换模型(Substitution Model),二是不同位点间速率异质性模型(Site Heterogeneity Model,即位点之间有没有替换速率的差异)。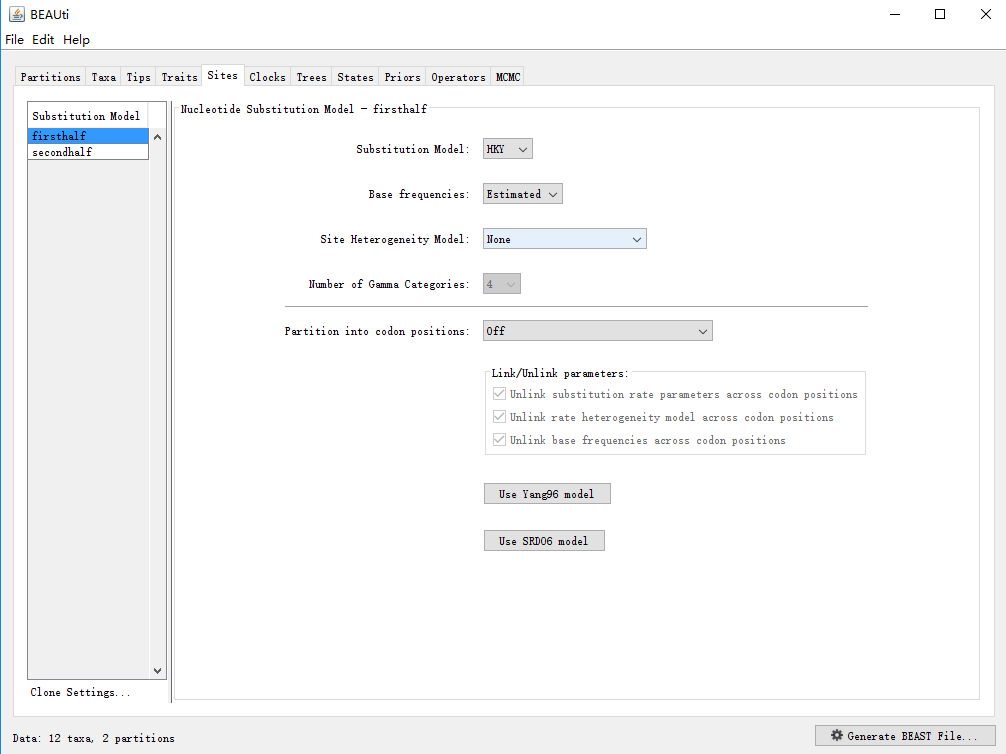 如图,如果是分区数据,可对每个分区设置模型及参数。这些模型一般是通过其他软件比如jmodelTest(核苷酸序列)或protTest(氨基酸序列),或者ModelFinder(核苷酸和氨基酸均可,推荐)测出。
如图,如果是分区数据,可对每个分区设置模型及参数。这些模型一般是通过其他软件比如jmodelTest(核苷酸序列)或protTest(氨基酸序列),或者ModelFinder(核苷酸和氨基酸均可,推荐)测出。
③关于替换模型及位点变异速率模型,这里不再啰嗦,前面的教程都提到很多,需要注意的是:
(i)当序列是编码区和非编码区的组合时,数据分区非常有用。
(ii)GAMMA位点变异速率的shape值,对于Gamma分布而言,其有一个关键参数,叫shape值。当shape值很小时,不同位点变异速率曲线呈L型,多数位点缓慢进化(小的替换速率),少数位点快速进化(高的替换速率);shape值较大时,不同位点变异速率曲线呈钟型曲线,多数位点进化速率相同,差异小。
(iii)针对编码区密码子分区这里: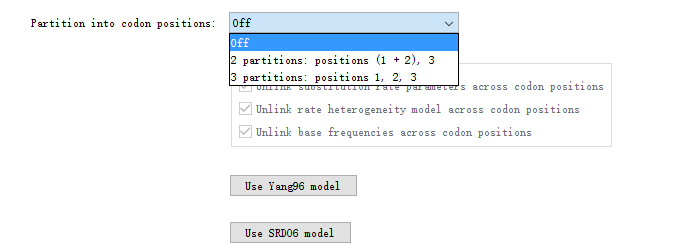
默认密码子分区是关闭的,“(1+2),3”的意思一共分成两个区,所有序列密码子的第一和第二个碱基在同一区,第三个碱基单独一区;而“1,2,3”的分区方式是一共分成3个区,密码子的每个碱基分别在不同区。
(iv)密码子的第1、2个碱基比第3个碱基进化速率小。
④关于SRD06 Model
点击这个模型后会自动设置相关参数,需要注意的是这个模型适合编码区序列。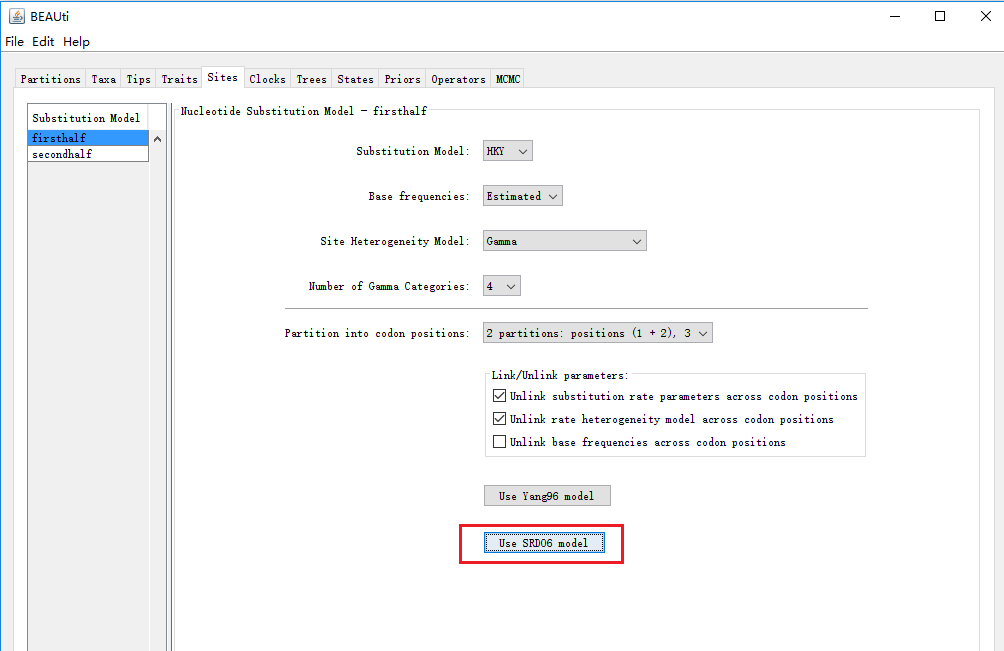
⑤固定平均变异速率(Fix mean substitution rate)
请注意:这个选项在Beast v1.4版本和2.0版本系列才有,v1.8版本和1.10.4里没有。
(i)当没有化石校准数据并且想要设置这个选项时,此选项是使用已知的替换(或突变)速率(substitution/mutation rate)来代替;
(ii)如果将此设置成1.0将导致内部节点的估算是以substitution/site为单位的。默认是1,当分析的目标是系统发育重建并且对系统发育的时间框架不感兴趣时,这通常是合适的。
(6)分子钟模型设置
这个菜单可以让你为树上不同分支之间的进化速率变化选择适当的模型,你选择的模型将用于估算每个节点的替换速率、taxon分类群的tMRCA(如果有的话)、treeModel.rootHeight(represents tMRCA for the root of the phylogeny)。
点击菜单栏“Clock”,如下: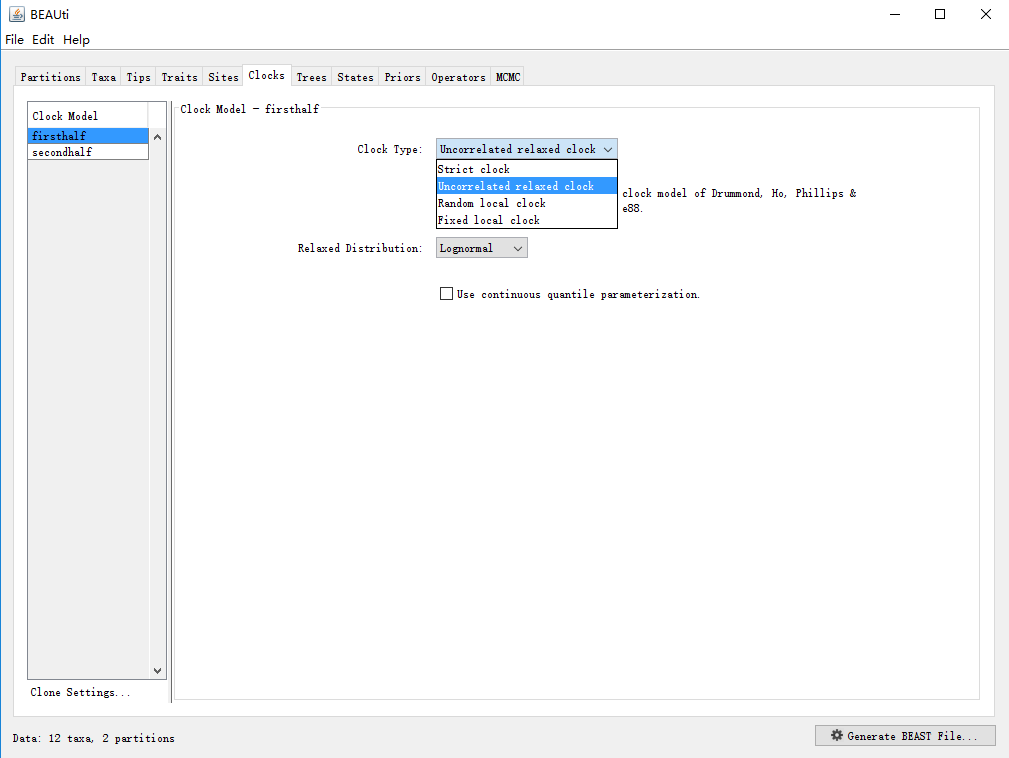
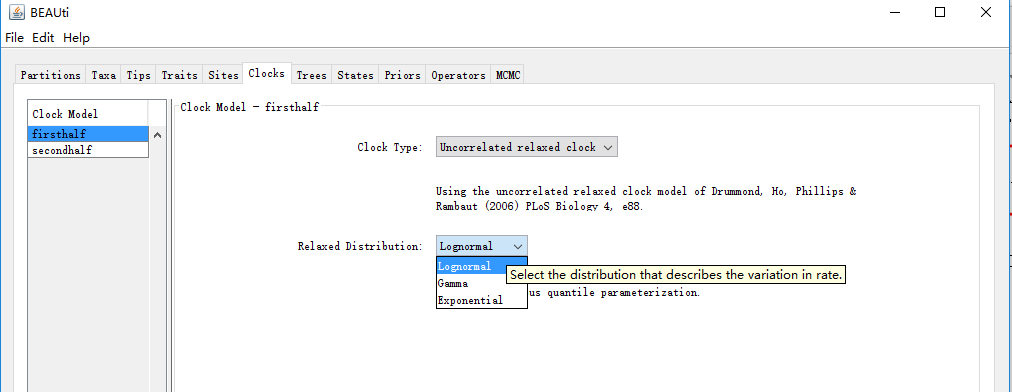
一共有以下几种分子钟模型:
① 严格分子钟(Strict Clock)。
严格分子钟采用全局 clock 速率且假定在树的谱系之间没有差异,即不同枝有着相同的进化速率。严格的分子钟通过以下任何一种方式来校准:
(i)指定一个变异速率。如果选择了“Strict Clock”,在“Priors”菜单栏下会有“clock.rate”参数,可以调节这个参数。
(ii)对“Priors”菜单栏的“tmrac”指定一个先验,或者设置“treeModel.rootHeight”值。
② 宽松分子钟(Uncorrelated Relaxed Clock),又分为 Lognormal(对数正态)、 Exponential(指数)、Gamma分步。宽松分子钟假定不同枝有着各自的进化速率,宽松的分子钟之所以称为“Uncorrelated”,是因为宽松分子钟在各谱系的进化速率和祖先之间没有已知的相关性(priori correlation )。
③随机局部分子钟(Random local Clock)。
④固定局部分子钟(Fixed local Clock)。
如何判断自己的数据适合宽松的分子钟还是严格的分子钟?(此处复制From高芳銮教授QQ空间日志教程)
方法一:可以先用uncorrelated relaxed lognormal clock配合默认参数配置个xml文件,MCMC链长不需要很大,运行结束后,将xx.log文件通过Tracer导入查看coefficientOfVariation值,如果该值接近0,说明数据适合严格分子钟,反之说明数据适合宽松分子钟。
方法二: 通过贝叶斯因子法(Bayes Factor, BF)来比较分子钟模型和树先验(tree prior)模型之间的组合的边际似然值(Marginal Likelihood),通过BF法分析后,哪个模型对应的LnL值越大,相对更优,此方法既可以确定分子钟模型,也可以用于确定哪种树先验模型更加适合自己的数据。具体也可以通过以下操作来确定:在Tracer 中同时导入两个模型的xx.log 文件,选中所有要比较的模型后,依次点击菜单Analysis—->Model Comparation,Analysistype改为Harmonic mean,Reps=100,最后根据获得的BF值(BF=Pr(D|M1)/Pr(D|M2)至少大于3,则说明M1>M2)范围判断模型间的优劣程度。
方法三(更新): 同样是采用比较分子钟模型和树先验(tree prior)模型之间的组合的边际似然值(Marginal Likelihood),但是不使用BF法,而是采用path sampling(路径采样)分析,相比BF法,path sampling更加精准。(参考文献:Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty)。
在Beast1版本里启用路径采样分析方法如下图:
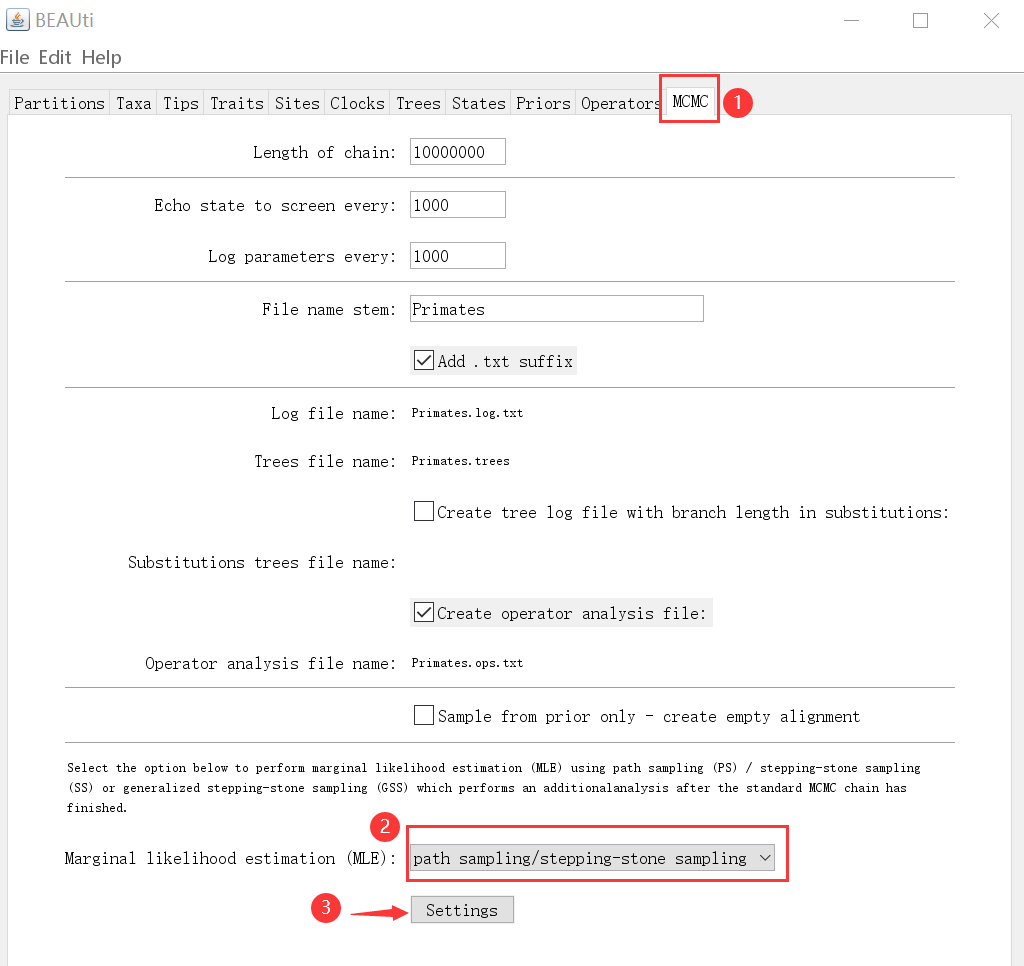
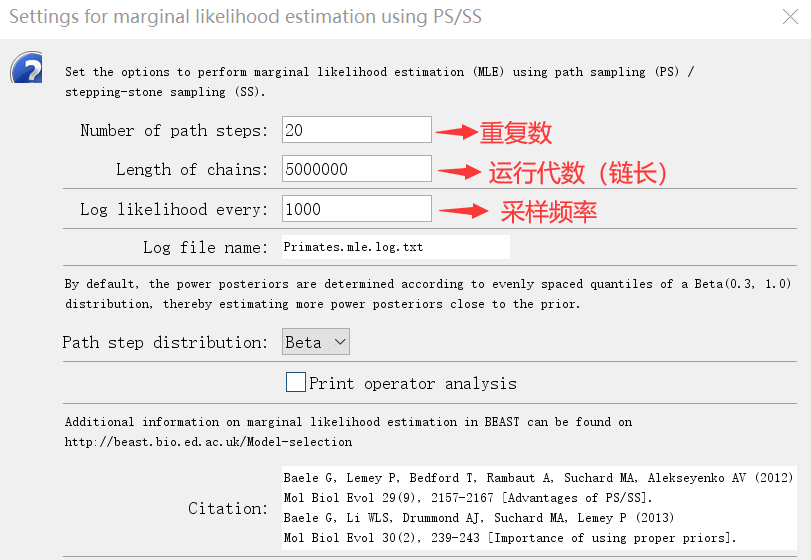
“Setting”里配置如下:
(7)Tree priors
树先验,该选项用于设置种群或者物种规模随时间的变化模型。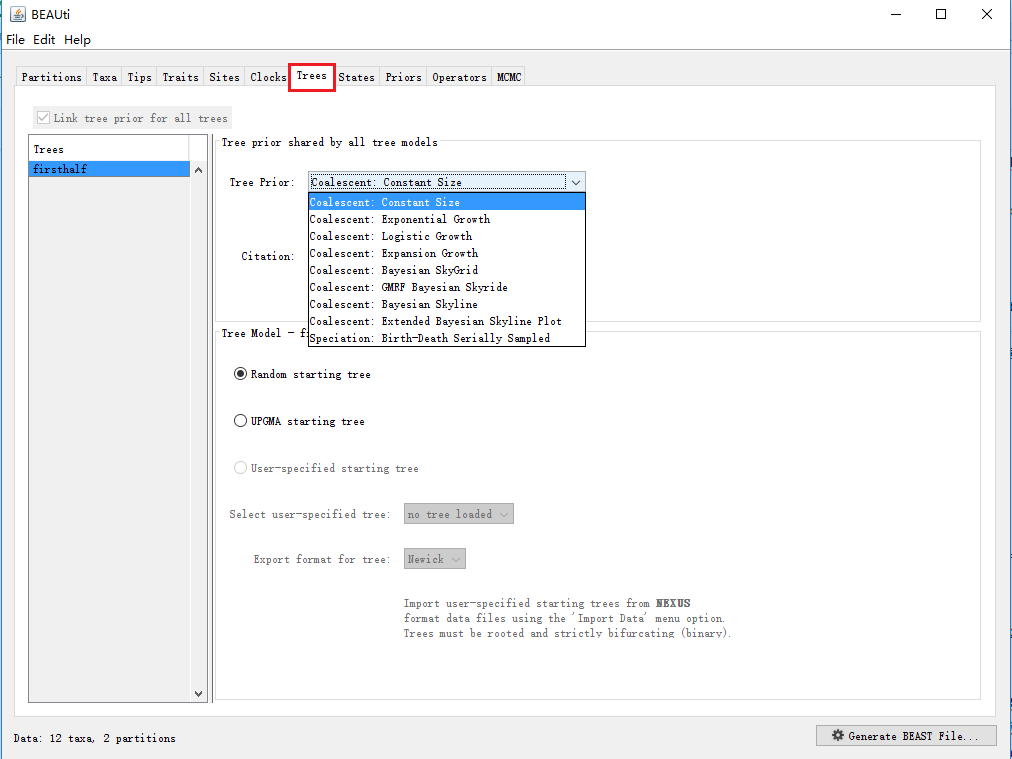 ① 如果序列是收集至种内随机交配的种群,在这种假设下,可以使用各种溯祖理论下的树先验来模拟种群大小随时间的变化。
① 如果序列是收集至种内随机交配的种群,在这种假设下,可以使用各种溯祖理论下的树先验来模拟种群大小随时间的变化。
(i)常见的种群规模随时间有三种增长方式,恒定大小/规模(constant size)、指数增长(exponential growth)、对数增长(logistic growth),具体用哪一种取决于你在分析时做的假设。
(ii)此外,还可以使用Bayesian skyline plot(BSP,贝叶斯天际线)种群增长模型,即随着时间(与世代间隔呈常数相关)的推移,计算有效的繁殖种群大小(Ne)。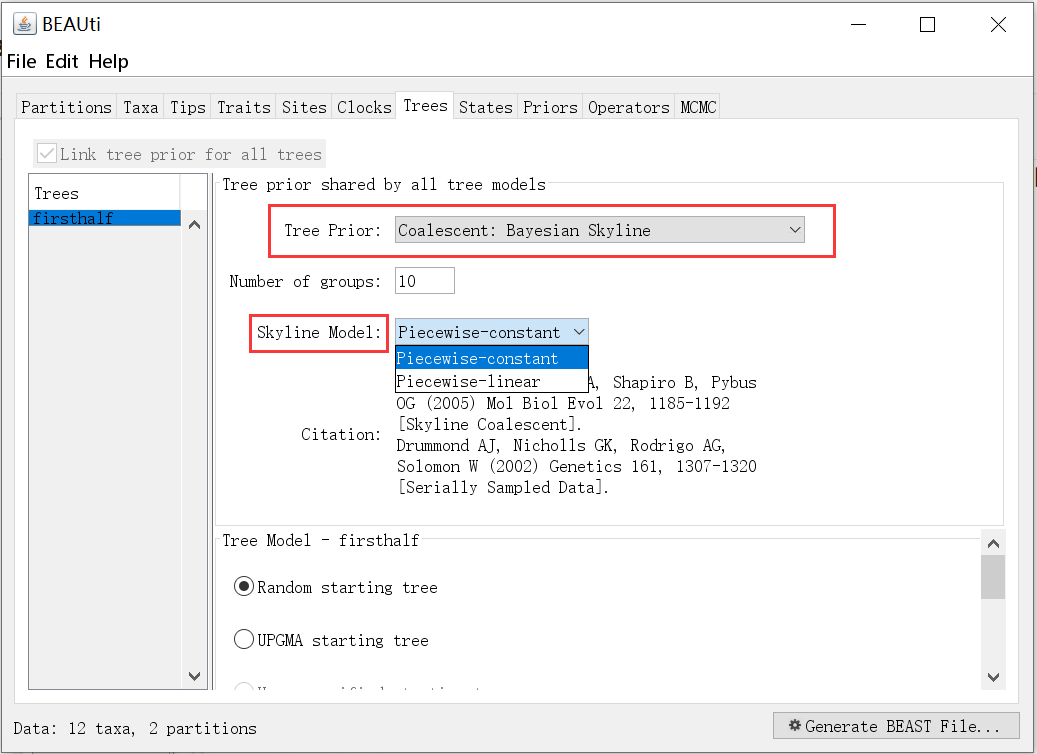
BSP使用的范围:序列数据中包含非常强烈的种群历史相关的信息,或者种群统计历史不是主要感兴趣的对象并且一个带有最少假设的灵活的溯祖树先验是可取时。这种基于溯祖理论的树先验只需要指定允许在种群历史中进行多少离散变化。
the BSP should only be used if the data are strongly informative about population history, or when the demographic history is not the primary object of interest and a flexible coalescent tree prior with minimal assumptions is desirable. This coalescent-based tree prior only requires you to specify how many discrete changes in the population history are allowed.
BSP模型会估算一个种群统计函数,这个函数具有一个指定步骤数,对每个步骤中的变化点和种群规模在所有时间的积分。
另外还提供了两个BSP的变异模型,即:
constant模型表示种群规模在变化点之间是常量,然后瞬间跳跃(population is constant between change-points and then jumps instantan)。
而linear模型表示种群规模在变化点之间呈线性增长或下降(the population grows or declines linearly between change-points)。
上面列出的所有种群统计模型都是树中节点年龄的参数先验。
②对于物种水平(species-level)的系统发育分析(序列来源于不同的物种),溯祖先验(coalescent priors)通常是不合适的,需要使用物种形成模型。建议使用Yule Process,这是一种纯粹的出生模型,所有的世系都有可能分裂成两个后代世系。Yule Process假定每个系谱都有一个指定的常量速率,通过“yule.birthRate”(the average net rate of lineage birth)参数来指定。yule先验参数(yule.birthRate)通常被认为是描述物种形成的净速率, 在这种先验下,分枝长度将以指数分布和yule.birthRate−1来被估计。
④具体如何选择哪一种合适的Tree priors,一方面取决于所用的数据(比如来源于相同物种用种群模型,或来源于不同物种用物种模型)。例如,来源于相同物种的不同种群数据,但是不知道使用恒定种群规模还是指数增长,还是BSP,可以做模型比较(上面第(6)部分已经给出方法)。需要注意的是,如果是进行发散时间定年和突变速率估算,则应注意树先验有可能影响突变速率估算。
关于Tree priors,笔者在目前已经发表的一些文章中也注意到,在病毒学研究中(注意这句话,后面说的都是病毒),即使是来源于不同物种的毒株序列,也经常见到使用种群相关的溯祖(Coalescent)理论模型(比如BSP等)。倒不是说用错了,根本原因在于,种群和物种从其定义来看,指的是能有性繁殖的生物,而病毒是无性繁殖。病毒的分类由ICTV根据一些保守基因的同一性来划分科属种,相比于动物这样的有性繁殖生物,病毒没有严格的种群和物种这样的概念,于是从广义上来看,即使是来源于不同物种的毒株序列,某些情况下,使用溯祖(Coalescent)理论相关的树先验模型,也有一定的合理性(个人拙见,有错误烦请告知)。
(8)Priors选项里部分参数介绍
设置先验优缺点并存。优点是比如可以将相关知识如已知的化石校准点或者已有文献里的一些分析结果,作为先验值纳入分析,而缺点则是先验如果设置不当可能会影响后验,进而影响最终的结果,设置不当有可能得到一个错误的结果,比如对进化速率,分歧时间等等这些值的估计。
在设置“先验”前,需要明白“先验”含义,以及它与“后验”之间的关系。(软件用户)通过给定一个先验(本质就是给定一个取值范围),随后mcmc从先验中进行抽样,得到后验值(即估算的结果)。有趣的是,Beast最终估算的结果是在你给的范围内。
这个时候会有人说了,这样岂不是可以人为最终控制结果?答案是肯定的,但是没办法,贝叶斯推断就是需要先验,感兴趣的可以去了解下贝叶斯推断的原理,这里不展开。
参数的先验设置是一门大学问,其设置是否合适,取决于使用这个软件的熟练程度,以及对自己研究内容以及前人的一些研究结果等等的基本了解,实在不知道怎么设置,可以使用默认值,但是依据笔者的经验,如果需要估算分歧时间,“进化速率”这个参数的先验请谨慎对待它,往往不太建议使用默认值,对它的先验范围也不要设置太宽,仅仅放在这里作为参考。
下面仅介绍下“先验”设置里一些参数的含义:
①clock.rate
这个参数是严格分子钟的速率参数,仅仅在严格分子钟模型下使用。该参数的单位是substitutions/site /unit time。如果该参数设置为1,那么进化树枝长度的单位是substitutions/site。如果进化树的内部节点是通过化石数据来校正的,且这些化石数据 的是millions of years ago (Mya,百万年), 此时clock.rate这个参数的单位是substitutions per site per million years (subs/Myr).
②constant.popSize
只有在菜单栏“Trees”里面选择了“constant size” , 菜单栏“Prior”下面才会出现这个选项。
This parameter represents the product of effective population size (Ne) and the generation length in units of time(τ) .If time is measured in generations this parameter a direct estimate of Ne. If clock.rate is set to 1.0 then constant.popSize is an estimate of Neμ for haploid data such as mitochondrial sequences and 2Neμ for diploid data, where μ is the substitution rate per site per generation.
③covariance(协方差)
这个统计值是用来计算宽松分子钟下的进化树里亲代分枝和子代分枝进化速率的协方差。
(i)如果这个值是正数,表明在你的系统发育树里,进化速率快的分枝旁边的分枝进化树速率也快。
(ii)如果这个值接近0,表明进化速率快的分枝旁边的分枝进化树速率慢。这也意味着这个系统发育史里没有很强的进化速率自我校准。
④exponential.growthRate
指数生长速率。只有在菜单栏“Trees”里面选择了“Exponential Growth”,菜单栏“Prior”下面才会出现这个选项。
⑤exponential.popSize
表示在指数增长下当前的种群规模。
⑥gtr.{ac,ag,at,cg,gt}
这个参数仅仅当你选择GTR进化模型时才会在“Prior”下面出现。即A↔C, A↔G, A↔T, C↔G and G↔T。
⑦hky.kappa
这个参数表示转换/颠换的比值(transition/transversion ratio)。仅仅当你选择HKY85进化模型时才会在“Prior”下面出现。
⑧meanRate
仅仅用于宽松分子钟,它的含义是整棵树上估计的每个位点的替换数除以估计的整棵树的长度(the estimated number of substitutions per site across the whole tree divided by the estimated length of the whole tree in time),表示单位时间内每个位点的平均替换数,它有着和严格分子钟里的clock.rate参数相似的含义。
⑨siteModel.alpha
GAMMA替换速率的alpha(或叫shape)值,仅仅当选择 GAMMA替换速率时才会在“Prior”下面出现。
⑩siteModel.pInv
表示不变位点比例(the proportion of invariant sites),范围是0到1,含义是数据中的某些位点从未经受任何进化上的改变,其进化速率是相同的。注意,不变位点比例,不是指不发生变异的位点的比例,而是指进化速率相同的位点的比例。
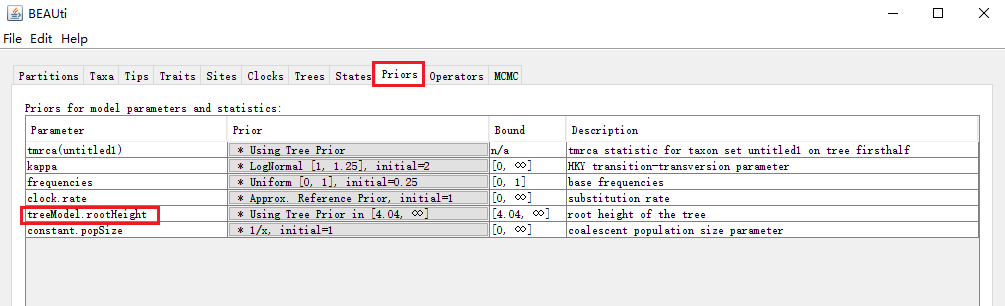
⑪treeModel.rootHeight
即“the age of the root of the tree”,这个参数表示树总的高度。这个变量的单位和进化树中分枝长度的单位相同(因为beast中的分枝长度已经代表时间),其最终值取决于进化速率的校准,或者加上节点分化时间的校准。
For divergence time estimation, calibrations are made by placing priors on the treeModel.rootHeight and tmrca(taxon group) parameters. For modern sequences, dates of divergence are set by creating priors on either internal nodes or the overall rate of substitution.
 ⑫ucld.mean
⑫ucld.mean
这个参数表示的是对数正态(lognormal)宽松分子钟下的平均分枝速率(mean of the branch rates)。这个参数和上面的meanRate有点相似但是不是相同的。如果 是第
是第 个分支上的速率,则ucld.mean是:
个分支上的速率,则ucld.mean是: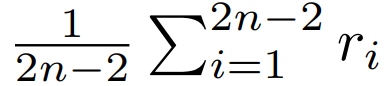
当你为对数正态宽松分子钟设置设置先验时,可以调节ucld.mean或meanRate这两个参数中的一个,但是注意不是两个都调(因为它们通过 参数高度相关)。
参数高度相关)。
⑬ucld.stdev
这个参数表示的是对数正态(lognormal)宽松分子钟下的标准差参数。如果这个参数为0,表示所有分枝的进化速率没有差异,言外之意这些序列数据实际上适合严格的分子钟。如果这个参数大于 1,则分支速率的标准偏差大于平均速率。
如果在软件“Tracer”里查看这个选项的频率直方图,如果频率直方图不和0接近且分枝间的进化速率存在差异,则表明这些数据适合宽松的分子钟,并推荐使用宽松的分子钟,示例如下(ucld.stdev=0.342):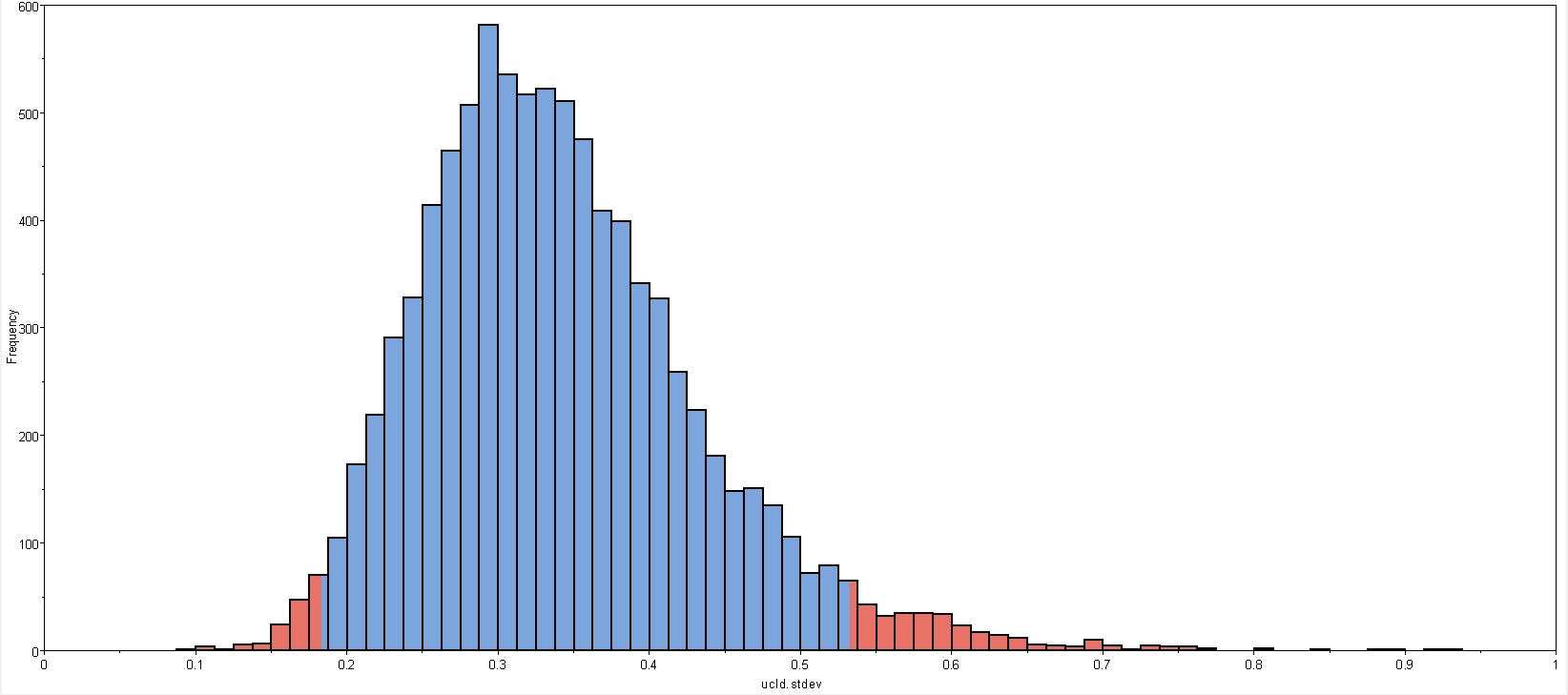
⑭yule.birthRate
这个参数是Yule模型里的谱系出生率(the rate of lineage birth)。如果 clock.rate是 1.0,那么这个参数估计的谱系出生数量(the number of lineages born)来自于parent lineage/substitution/site。
⑮tmrca(taxon group)
当你在“Taxa”选项里定义了分类子集,在“Prior”里设置“tmrca”这个参数或者设“treeModel.rootHeight”会起到同样的效果。
(9)Priors选项里的先验分布函数介绍
Beast里使用马尔科夫-蒙特卡洛进行抽样(采样),通过给定参数的先验分布(又叫取值范围),来估算参数最终的结果(也叫“后验值”),因此,需要一个函数来模拟或者描述参数的先验。常用的有均匀分布,正态分布和对数正态分布。
① 均匀分布(Uniform)
使用均匀分布作为参数的先验时,可以设置参数的最大值和最小值,初始值必须介于最大值和最小值之间。
② 正态分布(Normal)
这个分布先验允许参数从指定的正态分布中选择值,并具有指定的均值和标准差。
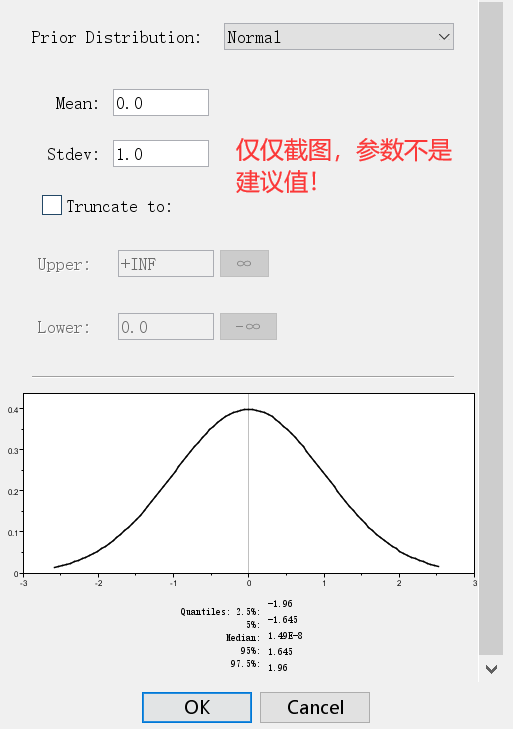
可以通过上面的曲线分布,获知你的参数取值的分布情况。
③对数正态分布(Lognormal)
这个分布先验允许参数从指定的对数正态分布中选择值,并具有指定的均值和标准差(以对数单位)。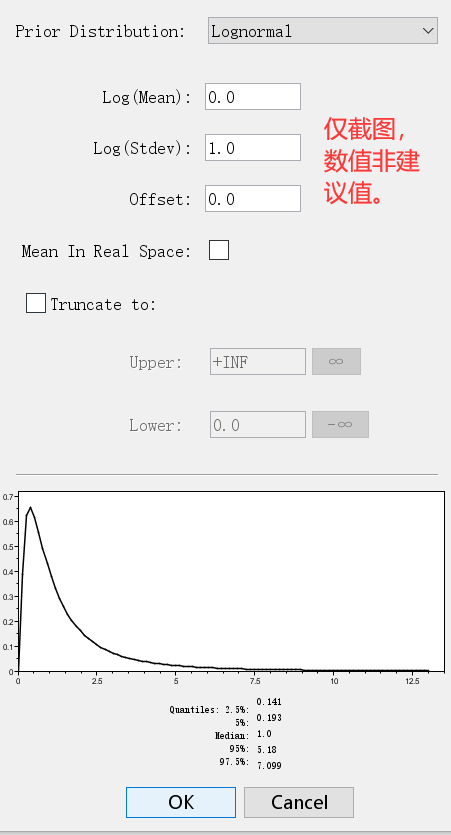
此时填入的值是经过对数化的。比如Mean这个值,你参考其他人的研究来填这个值,如果其他人给出的值是真实值,你如果选择了对数正态分布,这里需要先取对数后,再填进去。
当然,如果你觉得做对数计算麻烦,也可以选择填入真实值(没有取对数的),不过需要先勾选“Mean In Real Space”,之后可直接填入真实值 。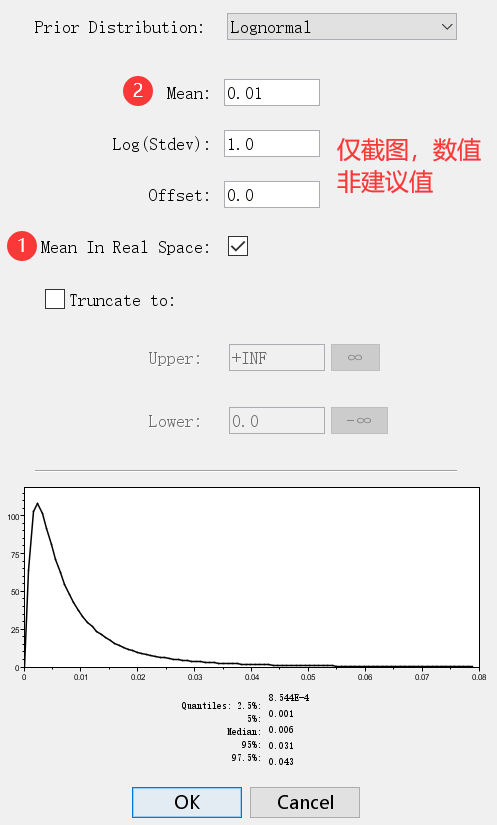
(10)Operators选项
①Tuning
更改这个选项下的数字将更改移动的大小,这将影响MCMC算法接受该更改的频率。
②weight
The weight column specifies how often each operator is used to propose a new state in the MCMC chain。
③Auto-optimize
The “auto optimize” option will automatically adjust the tuning parameters of operators as the MCMC algorithm runs,建议选择这个。
(11)MCMC 选项
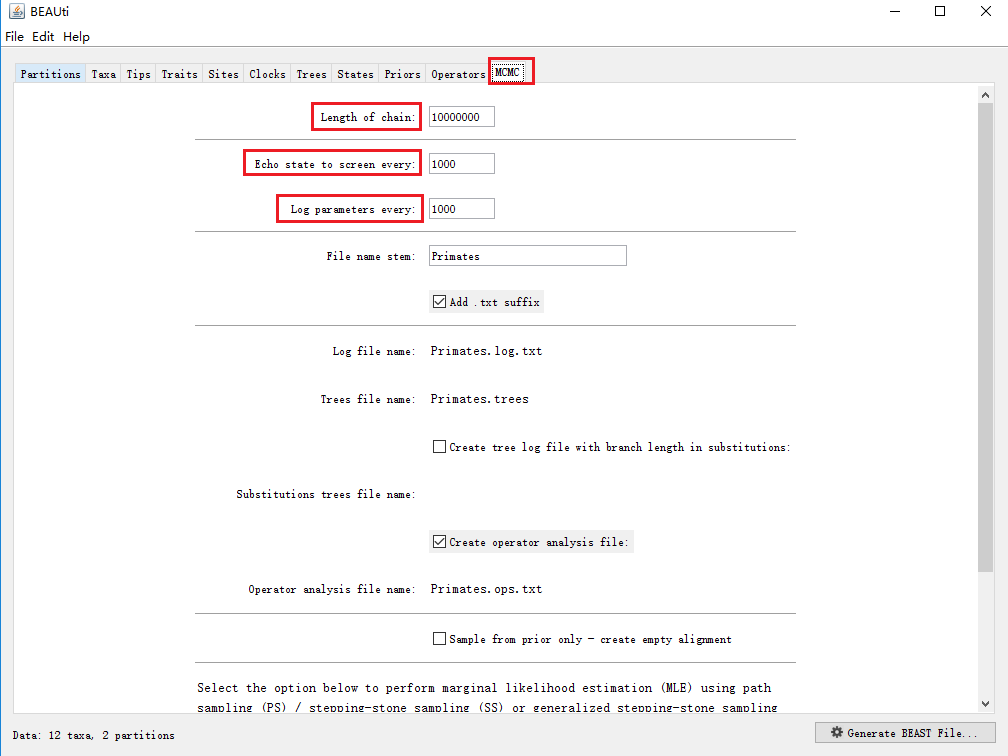
① Length of chain
指的是MCMC分析运行的总代数。可以通过以增加代数来提高ESS值(Effective Sample Size)使参数收敛。同时,增加采样频率也可以提高ESS值,但是过于采样频繁也不会提高ESS值。
那么对于不同条数的序列,总代数设置多少合适?一个粗略地计算是:总代数=3000 x 序列条数的平方。比如你有100条序列,总代数至少设置为30,000,000代;同理200条序列,总代数至少设置为120,000,000代。
② Log parameters every
多少的马尔科夫链的参数记录在日志中,表示采样频率,即多少代生成一个样本。这个值的设置受Length of chain影响,最主要的一点需要保证最后的样本最少得有10,000个。
样本数量=Length of chain(总代数) ÷ Log parameters every(采样频率)
③ Echo state to screen
将状态摘要输出到BEAST控制台窗口的频率。
④ The (subst)tree
The (subst)tree file will be a tree file with the branch lengths in units of substitutions.生成带有枝长的树。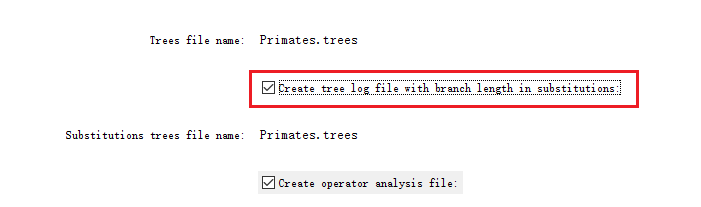
(12)生成XML文件
所有参数设置完毕后,点击下图红色方框按钮,生成用于后续BEAST运行的XML文件。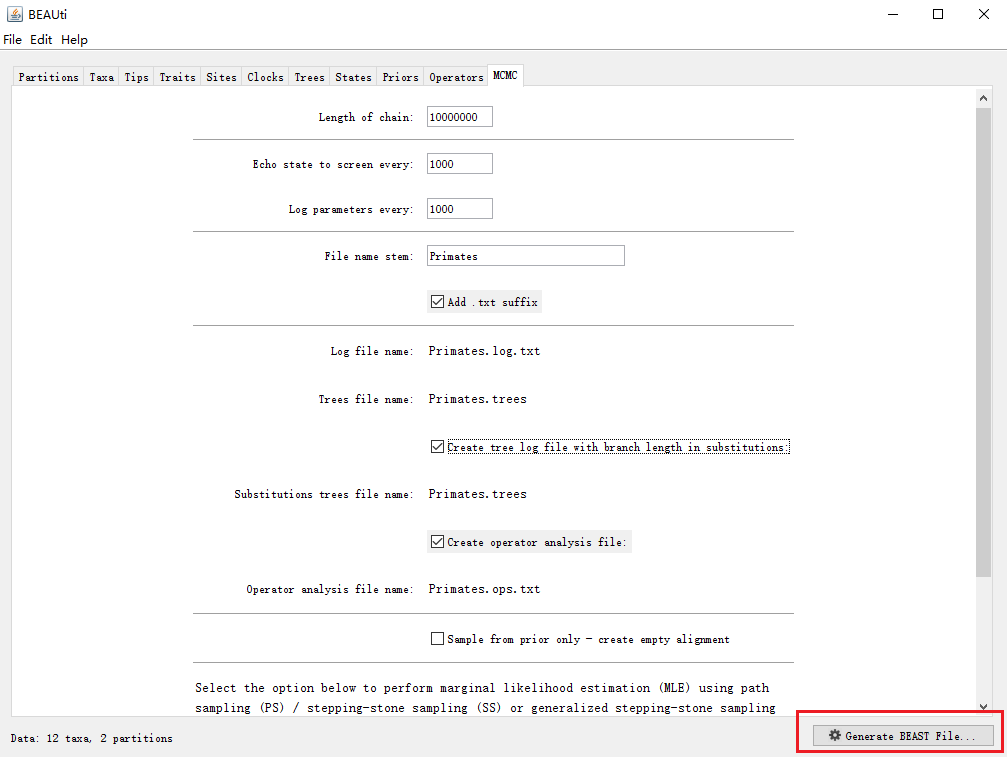
2. BEAST
(1)导入XML文件
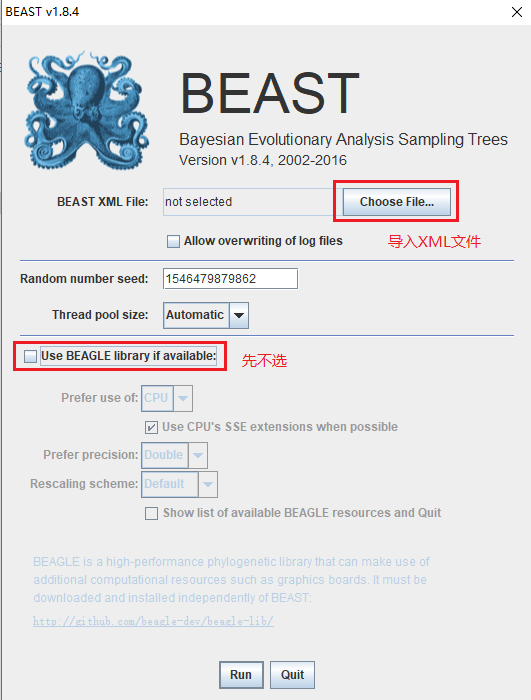
(2)注意
(i)BEAGLE library 的运行需要你安装一系列运行环境,它的功能是充分利用计算机资源来跑数据,如果你的计算机处理能力还可以,比如内存还算大,这个完全可以不选。需要注意的是,有些分析强制(必须)使用Beagle, 这里不拓展。
相关BEAGLE library知识及安装:
①http://beast.community/beagle#running-beast-with-beagle
②https://ericmjl.github.io/beast-gpu-tutorial/
③Linux系统上安装Beagle教程:https://www.yuque.com/wusheng/gw7a9p/sq0sqo
(ii) 如果你在软件BEAUti的MCMC选项里选择了生成带有枝长的树(The subst tree),当BEAST运行结束后,至少会生成下面4个文件: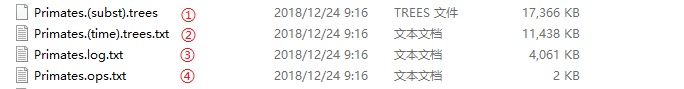
第①个文件是带有枝长的树文件,第②个文件是不带枝长的树文件,第③个文件是日志文件,第④个文件是相关的参数设置记录。
3. Tracer查看参数是否收敛
参数收敛的标准是查看各参数的ESS值是否大于200,ESS>200才收敛。将上述第③个日志文件(Primates.log.txt)导入到软件Tracer中,如下: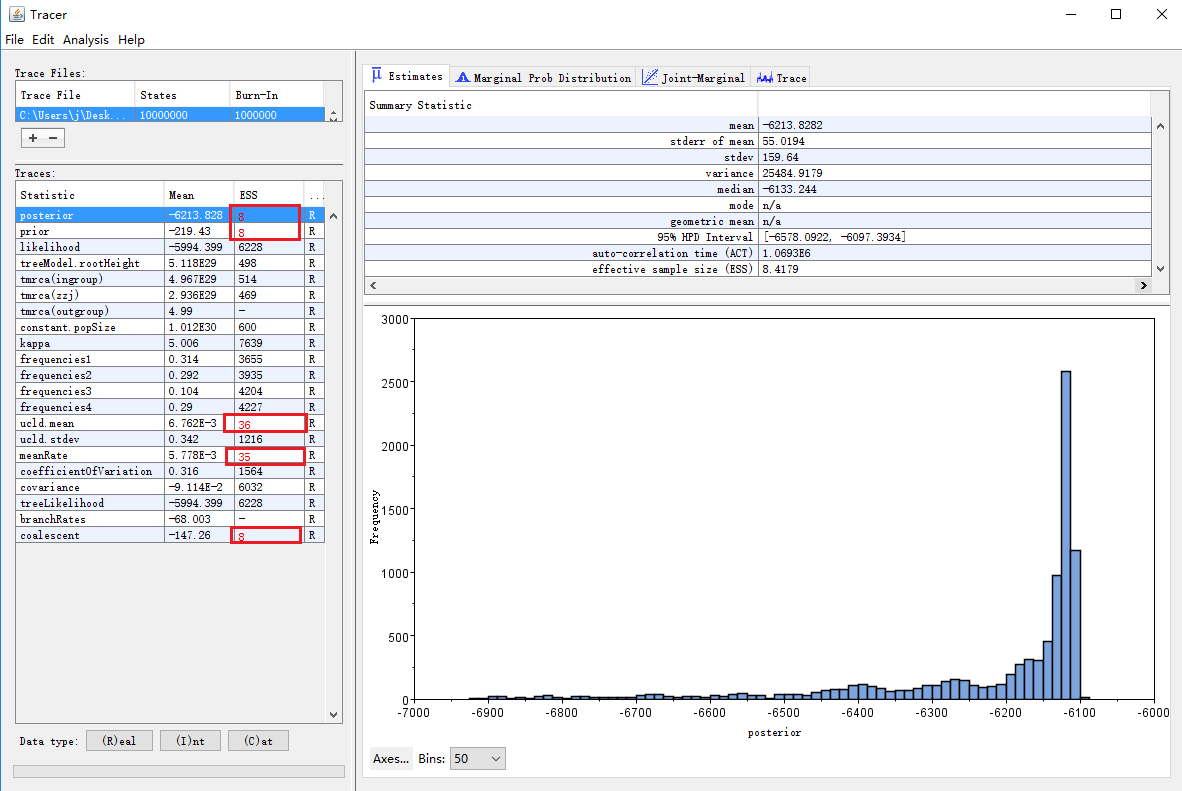
图中ESS值小于200的参数,即为没有收敛的参数。
对于提高参数的ESS值,官网给出的处理策略是:增加代数,或增加抽样频率,或合并多个独立运行链的结果。
笔者在实际使用中发现,提高整体参数的ESS值,在保证10000个以上样本的前提下,最好用的方法是:样本数先不变,增加抽样频率,将默认的1000调整为2000或5000或者1万等(此时运行总代数也会随之增加),若再不收敛,最后运行多个独立链,一般为2-4个,再合并多个独立运行链的结果。
如果最终还是有个别参数难以收敛,则重新配置XML文件,在“Operators”菜单里增加该参数的weight值,一般加大为原来的10倍。
4. TreeAnnotator获取树
① 打开软件TreeAnnotator,如下图1所示:
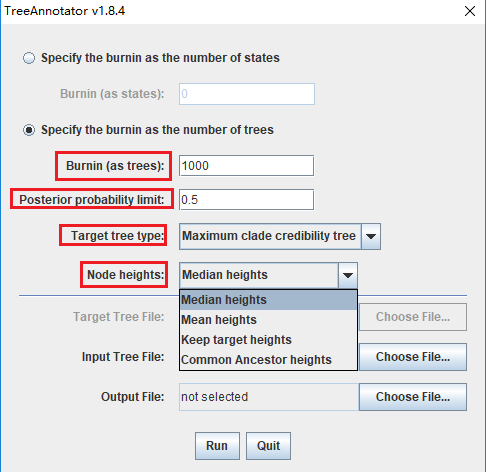
图1 图2<br />②** Burnin(as trees)指的是舍弃的样本数(树数),Burnin(as trees)=总代数 ÷ 采样频率 x 舍弃样本比例。如果舍弃样本比例取10%。**对于本文的示例数据,由于总代数设置为10000000、 采样频率为1000,故舍弃样本数设置为1000(图2)。<br />**Burnin(as states)**,舍弃的状态数,比如设置了1000万代,舍弃比例为10%,那么舍弃状态数则为100万代。<br />** Posterior probability limit 意思是后验概率低于多少的数值不在树枝上显示,我们一般设置为0.5。**(图2)<br />** Target tree type即想要建的树的类型,默认为MCC树(最大可信进化分枝树),如果想要其他类型的树,可选择第二个选项,并指定一个类型的树文件。**(图2)<br /> **Node heights:即节点高度的类型,可以选择中位数或者平均值等等。推荐使用默认的中位数,因为均值容易受到极值的影响。**
5. FigTree查看树及美化
将上述TreeAnnotato运行结果生成的MCC树文件(如果你选是的生成MCC树)用软件FigTree打开,如图: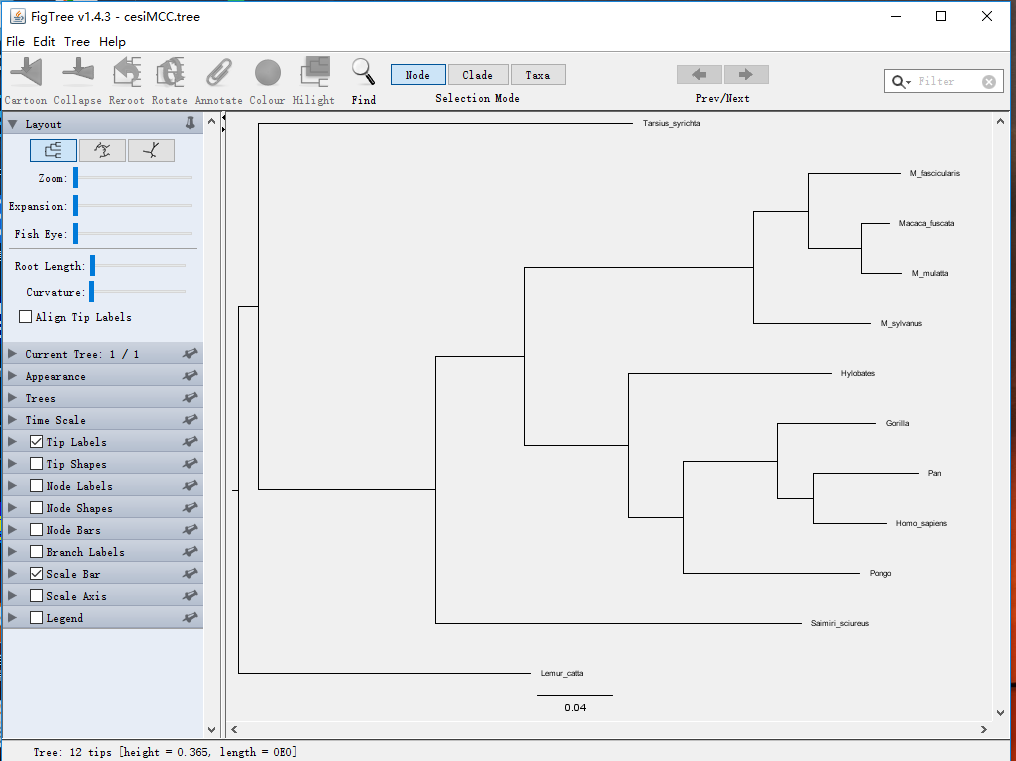
(1)将分枝按进化速率标色
① Appearance→Colour→rate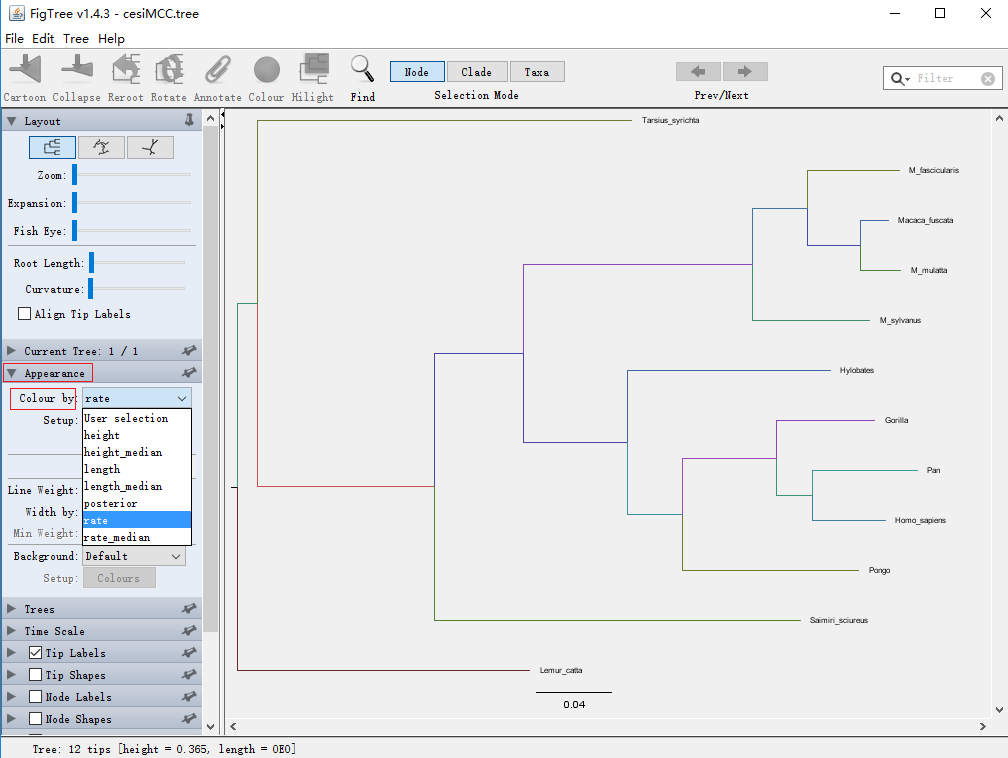
②导入速率比色卡
Legend→Attribute→rate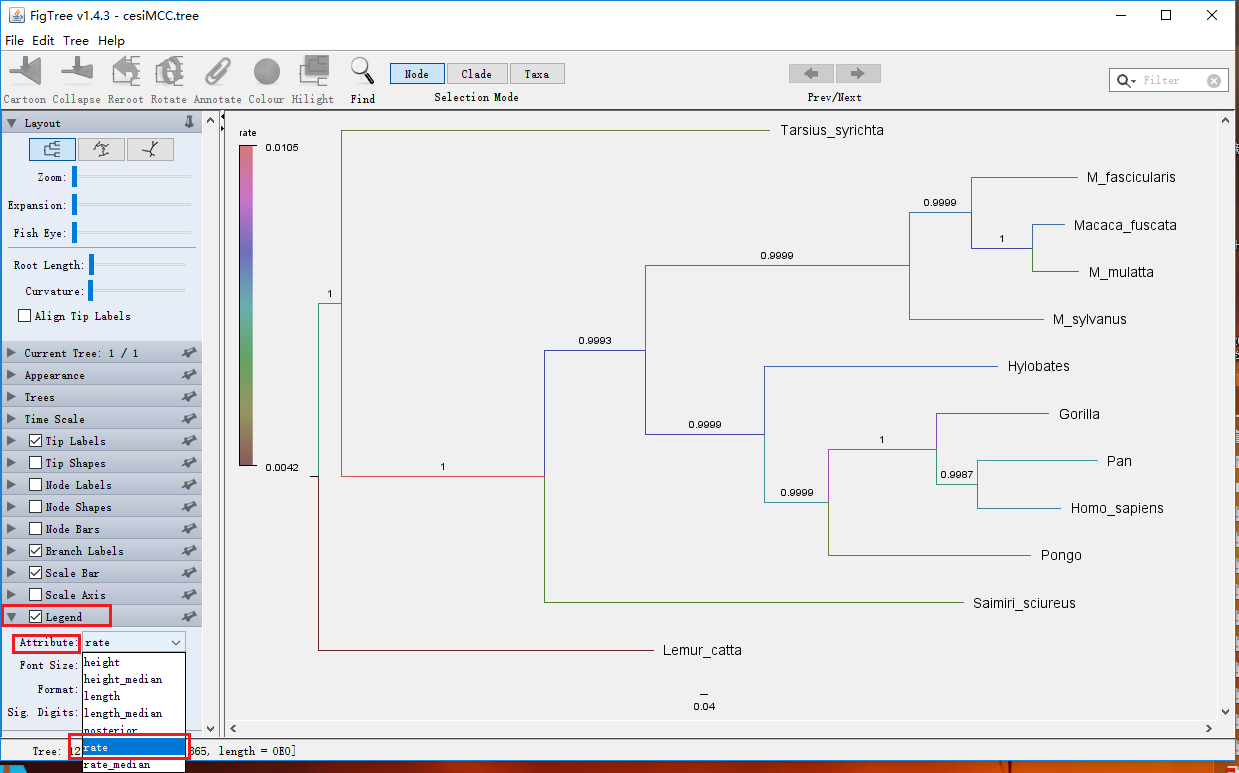
(2)修改字体大小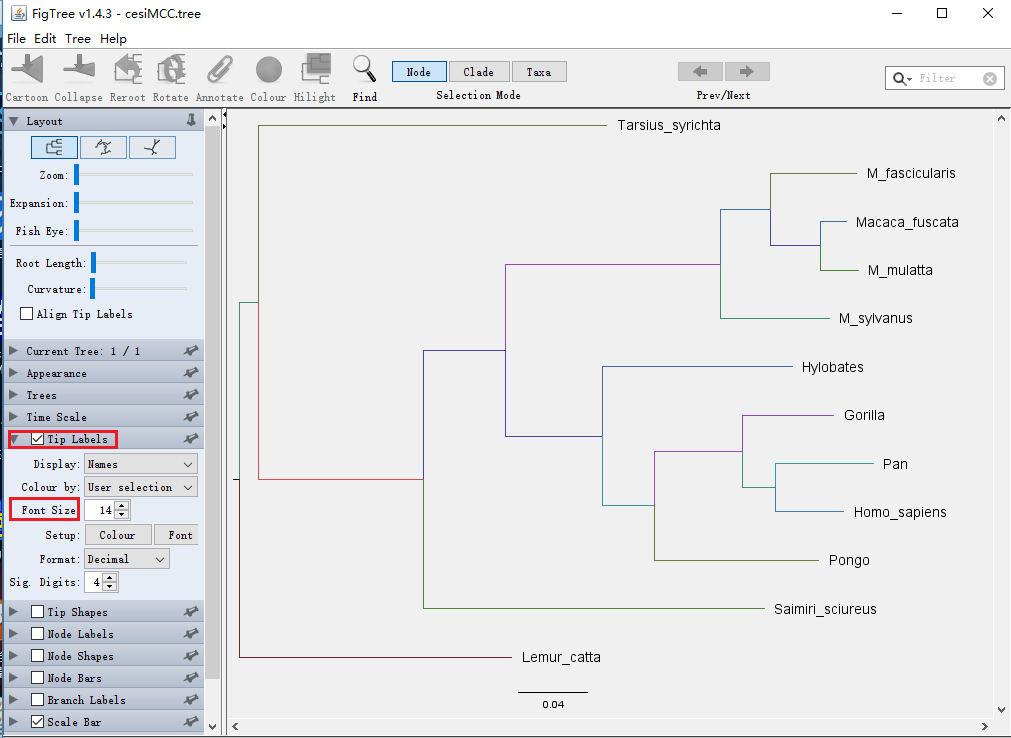
(3)以95%HPD作为节点标尺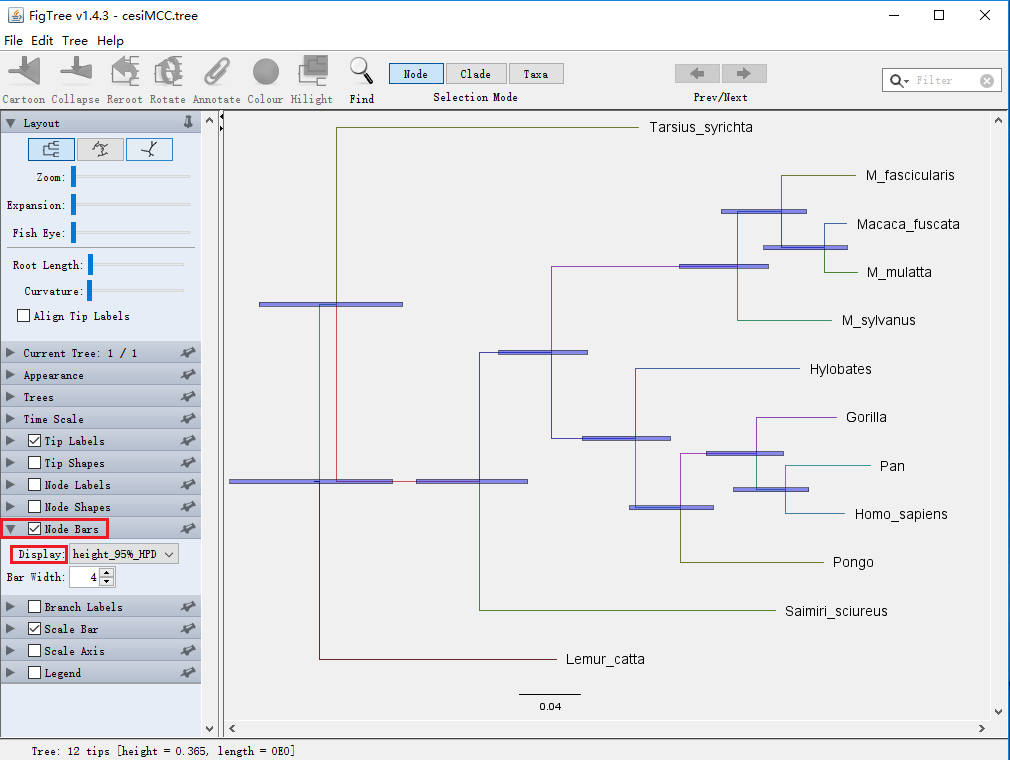
(4)以采集时间为标尺
①第一步:先勾选Scale Axis,再勾选Reverse Axis,如下图:(请点击放大更清晰)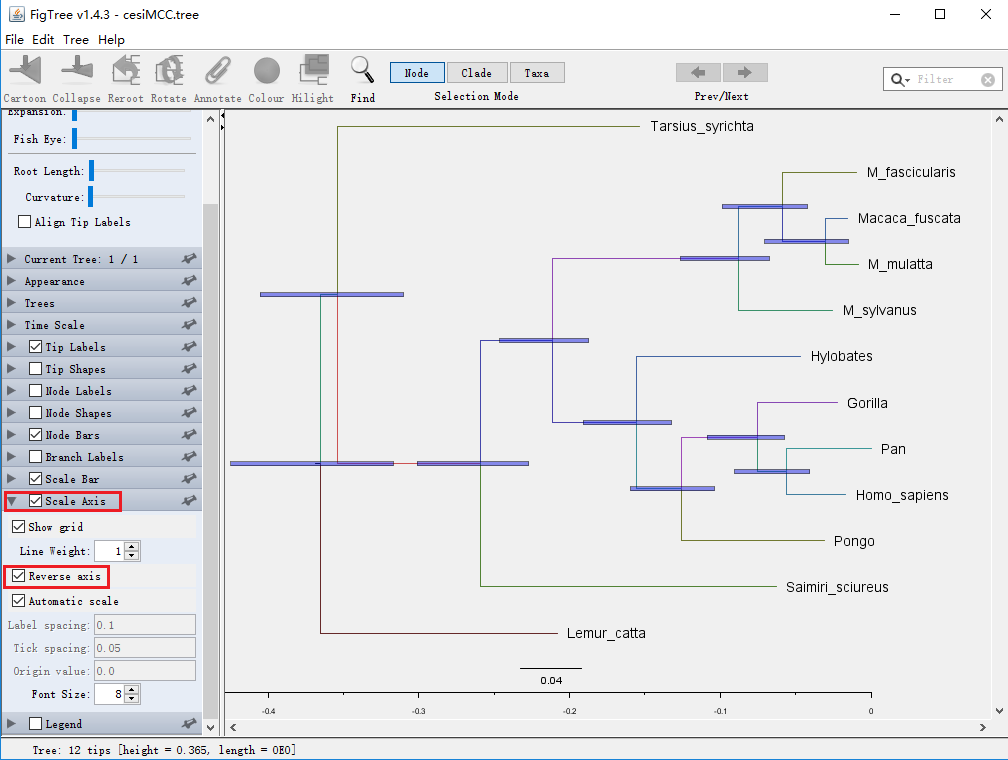
②第二步:将Time Scale 里的Offset by填上你用于分析的序列的最近日期。
想将图中的另一标尺如上图中的0.04去掉怎么处理?
这个简单,将Scale Bar前面的的√去掉就行。效果如下图: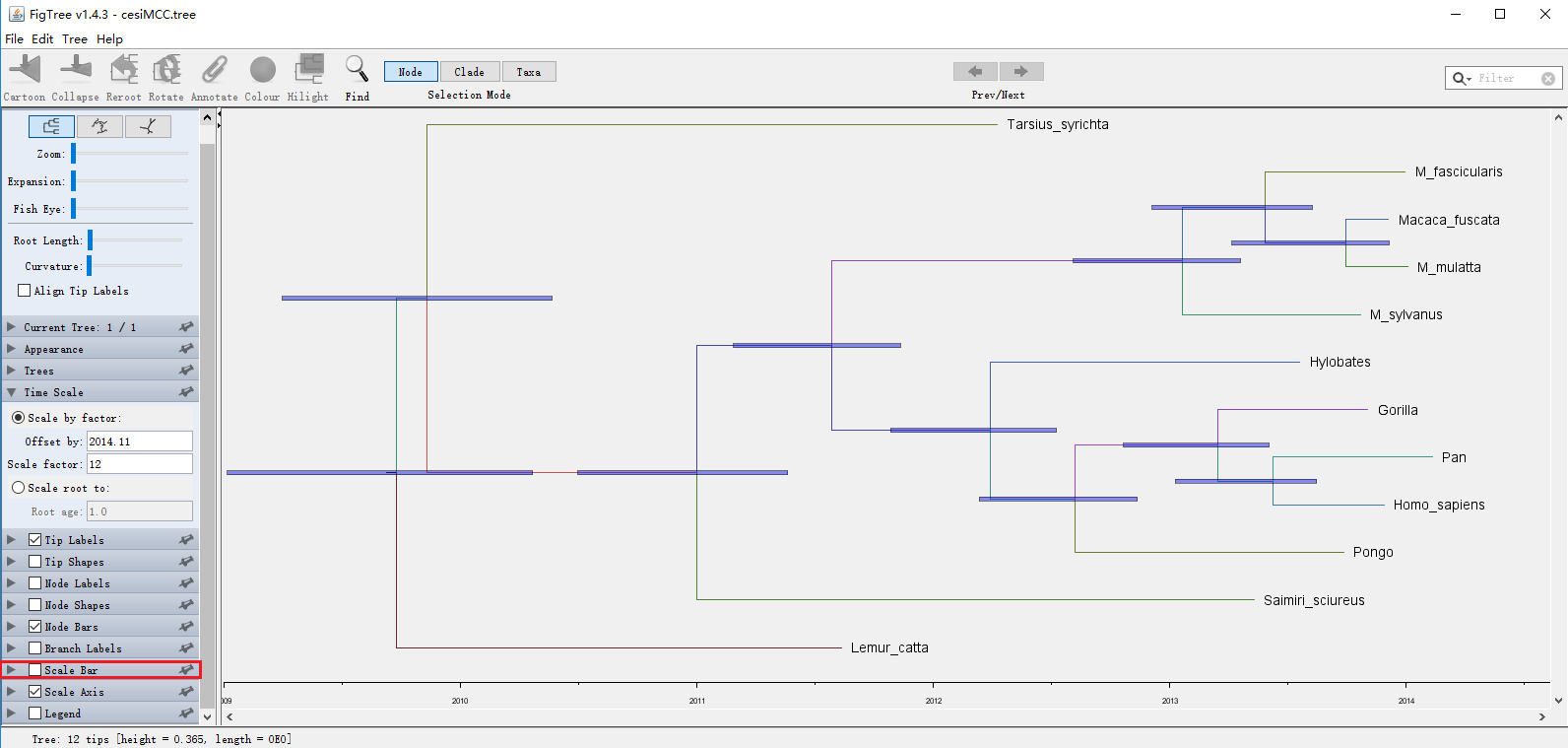
(5)显示后验概率
建议将后验概率显示在枝上,先勾选Branch Labels,再将Display选择为posterior,如下图(一棵带有时间标尺的MCC树):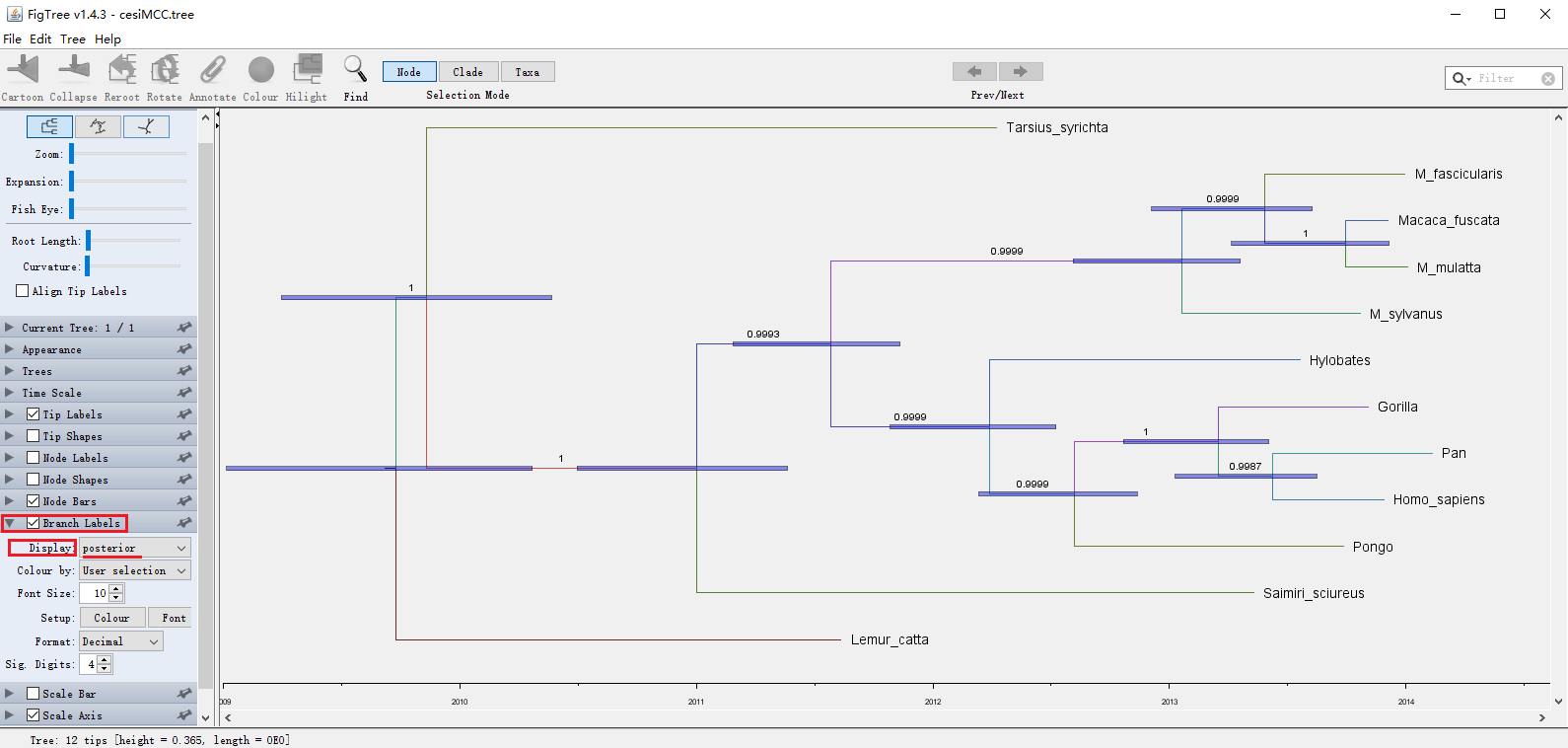
(6)显示节点年龄
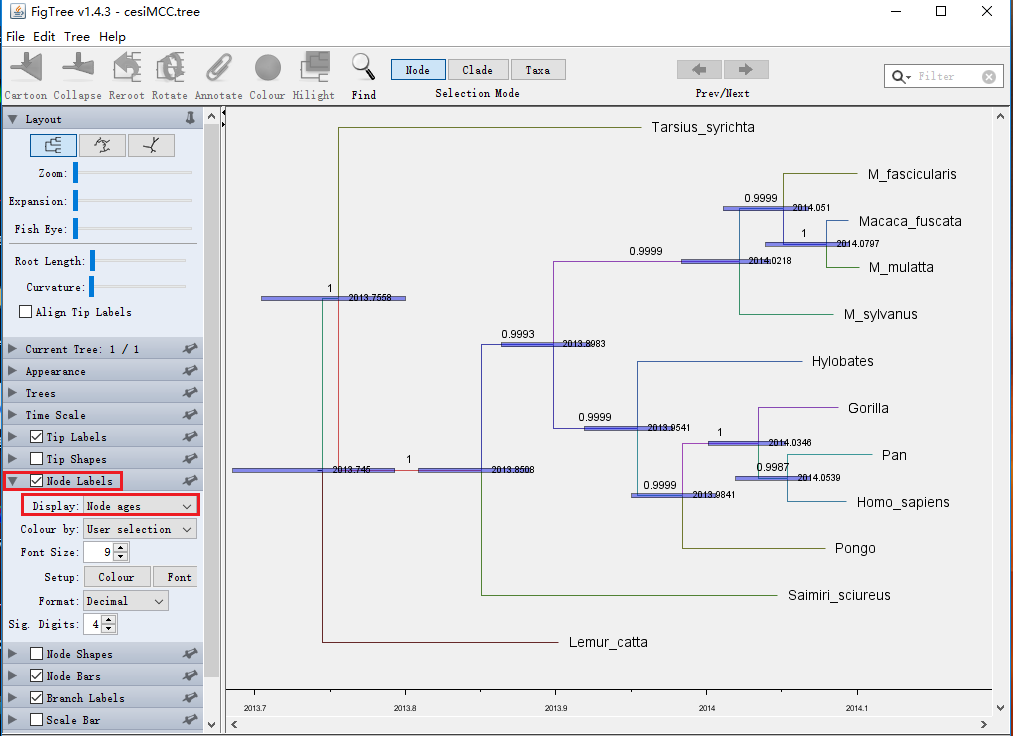
(7)注意
对于 Appearance这个选项下面的子选项还有许多意外的功能,请多多尝试。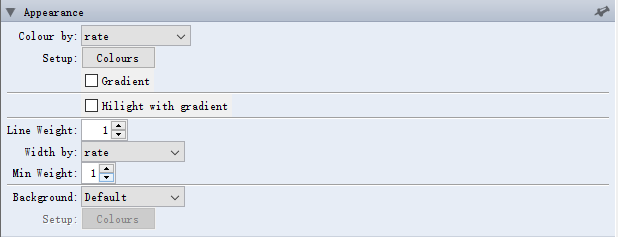
二. 构建带有时间标尺MCC树示例
1. 序列数据的准备
(1)先将序列整理.fasta格式的文件
格式如下图,如果想构建带有时间标尺的MCC树,还需要在序列名称上带上时间标签为后续做准备。
(2)将.fasta格式的文件转换成.nex文件
BioAider里的Seqformat Convertor序列格式转化功能:<br />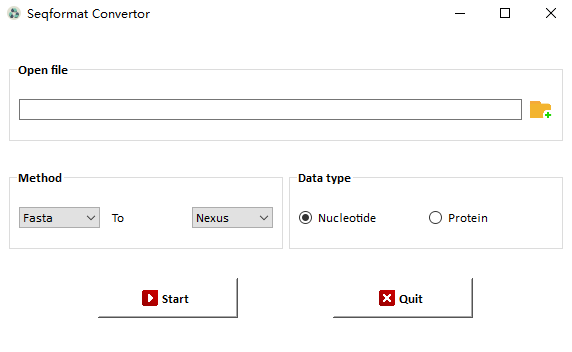
(3)查看.nex文件开头信息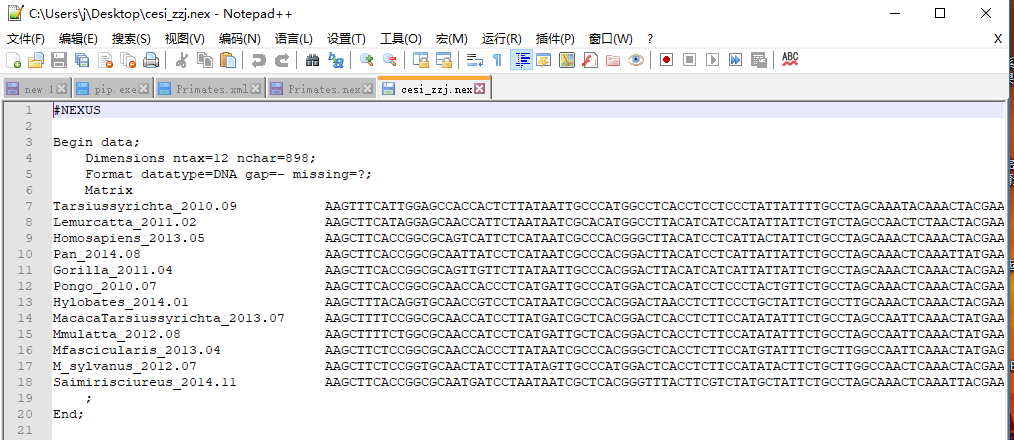
由于上面我们的序列排列为顺序式(sequential),为了和交互式排列进行区别(interleave)故推荐在下面位置加上一个代码 interleave=no: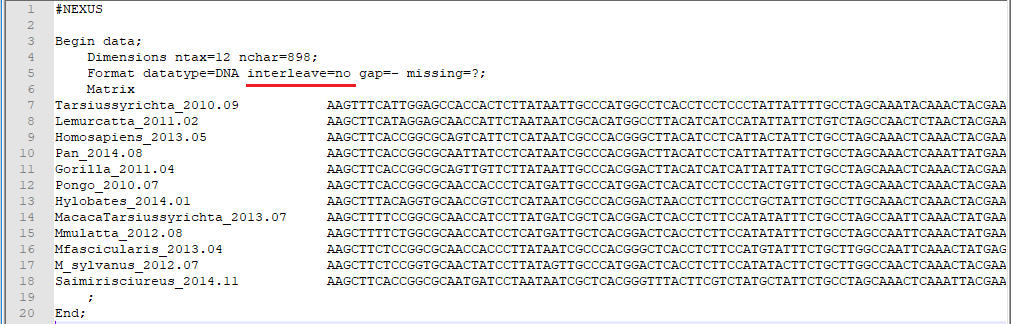
请注意:如果是分区数据建树,则需要在代码块最后部分加上分区信息。本示例不做分区数据建树。
2. 将.nex序列文件导入BEAUti
File→Import Data,选择.nex格式的序列文件。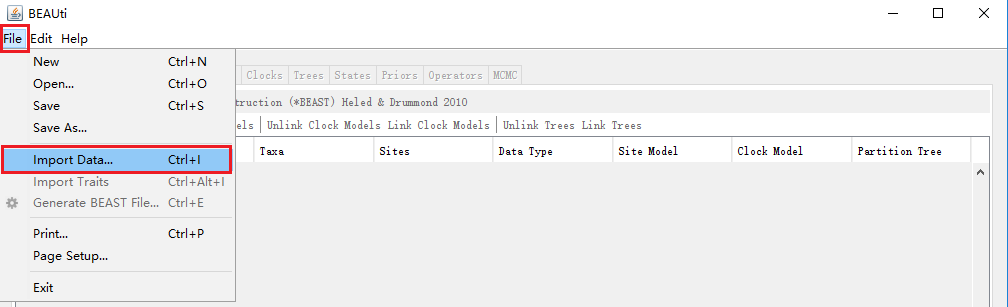
3. Taxa设置
这个设置并不是所有分析都需要设置,有需要才设置,本示例不设置这个。
4. 采集时间设置
①在将这些病毒序列的采样时间进行分子钟校准前,需要先判断分析数据集中是否有足够的时间信号(Temporal signal)可用于校准。
常用的判断方法有3种:
(i)做“根到节点的遗传距离图”,可以通过软件Treetime(推荐)或者TempEst完成,这种方式比较快速省事且非常适合快速进化的病原(比如病毒),具体软件使用方法请参考之前笔者分享的这个两个软件的教程。
(ii)使用日期随机化检验(date-randomization test),详细请参考下面附件里的教程:
日期随机化检验(分子钟校准前).pdf
笔者曾经用过这种方法,说实话,这种方式计算量也大很慢,之后再也不想用这种方法。
(iii)最近新出了一个软件叫BETS,用来检验数据集的时间信号,据说效果不错,笔者尚未研究,大家可以去试试。
②笔者假设(请注意这个词)示例数据具有较好的时间结构 (即基本符合严格分子钟) ,我们通过点击“Tip”设置序列采集日期,具体设置方法请见前面部分。(Use tip dates→Guess Dates→)
5. 进化模型设置
(1)在这一步之前,请先用模型软件比如jmodeltest/modelfinder(针对核苷酸序列)或者prottest/modelfinder(氨基酸序列)计算出序列的最适模型。
本文假设所用模型软件是jmodeltest同时基于BIC标准且序列最佳模型是下面:
特别注意:①R(a)即Rate [AC];R(b)即Rate [AG];R(c)即Rate [AT];R(d)即Rate [CG];R(e)即Rate [CT];R(f)即Rate [GT],因为新版的jmodeltest在这里全部简写了。
②gamma后面的数值0.7340即gamma的shape值或者叫做alpha值。
③p-inv即不变位点比例(the proportion of invariant sites)。
①②③里的这些参数对于Beast v1.8是在菜单栏“Priors”里面进行设置,后面会提到;而如果是Beast v2.0系列,则直接在模型这里就可以设置。
则软件里的对应的模型参数设置为下面: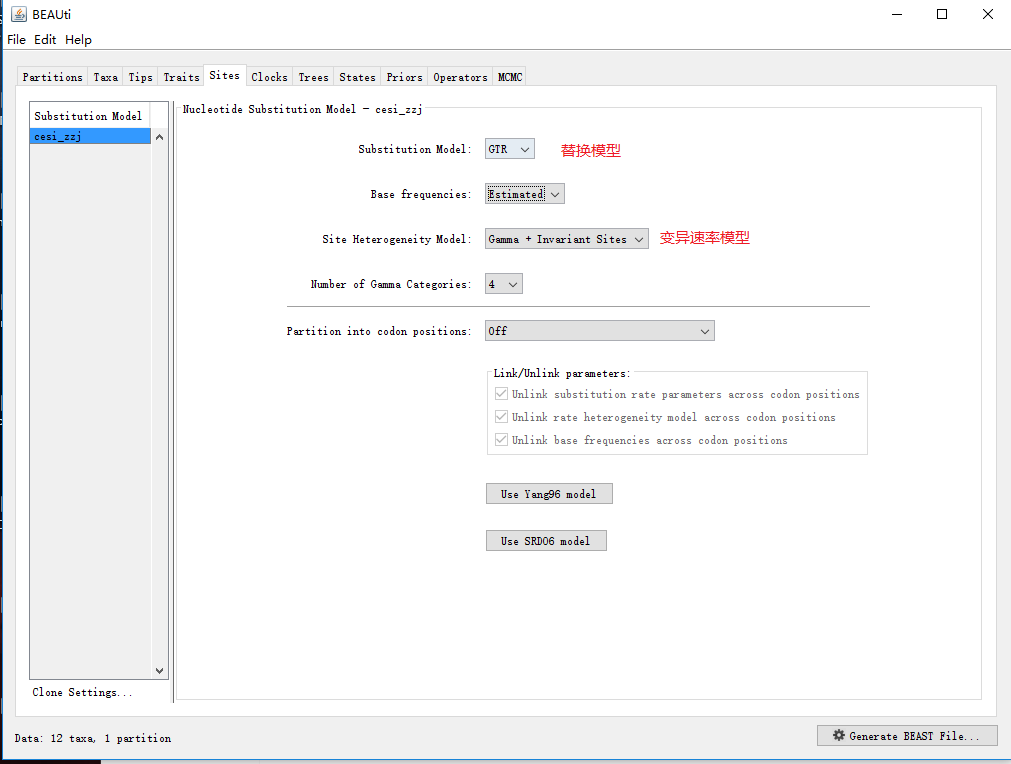
(2) 如果模型软件测出的进化模型,在这里没有对应的选项怎么办?
那么可以先在这里选一个模型,后续生成XML文件后,再在XML文件的模型那部分的代码进行相应的修改。
为了方便理解,先将上述示例进化模型(GTR+GAMMA+I)在XML文件里对应的代码部分提前放在这里: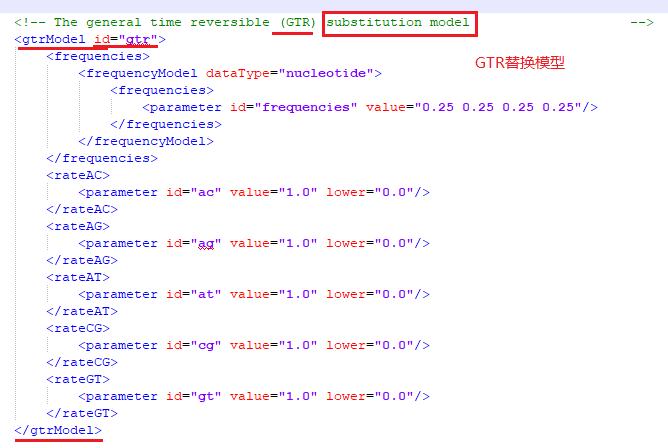

但是,你会发现比如果是其他核苷酸替换模型,比如HKY模型,还会有其他地方不一样:
所以, 如果需要自定义模型还是比较复杂的,可能定义得不对,各进化模型之间的相似点和区别请参考笔者转载的一篇文章“替换模型速查(IQ-TREE)”或者见下附件:
Setting DNA substitution models in BEAST.docx
笔者的建议是,先看看你的进化模型和BEAUti里已有的进化模型哪个最接近,再用BEAUti里最近的那个进化模型先配置个XML文件,最后再对XML文件里的进化模型部分的代码进行微调改成你需要的那个进化模型。
6. 分子钟模型设置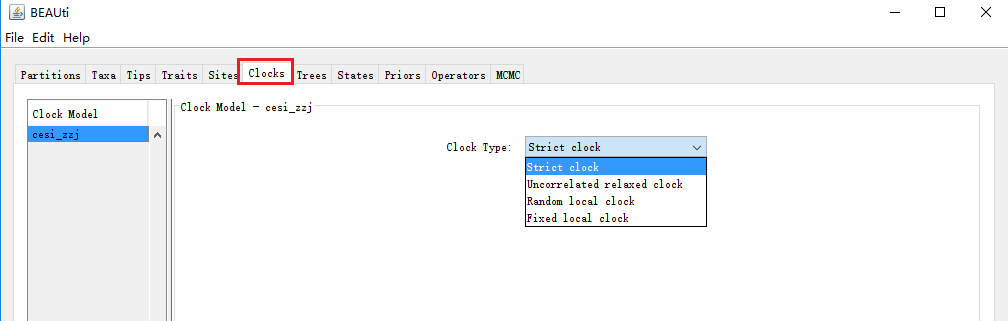
(1)如何判断自己的数据适合宽松的分子钟还是严格的分子钟前面已经给出方法。
(2)本文假设你已经完成(1)的步骤,且数据适合宽松分子钟,则参数进行如下设置: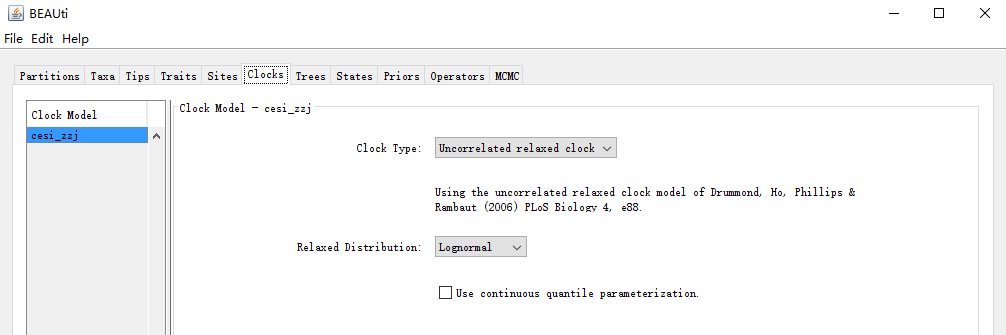
7. 树先验设置
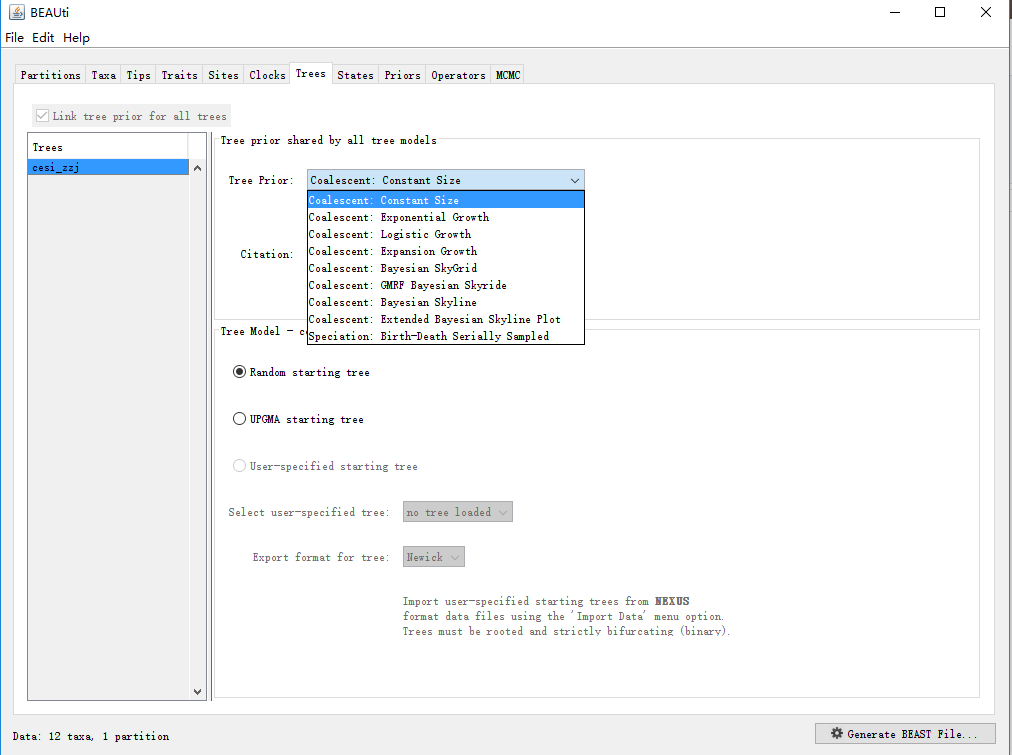
8. 先验部分参数设置
点击菜单栏“Priors”,如下: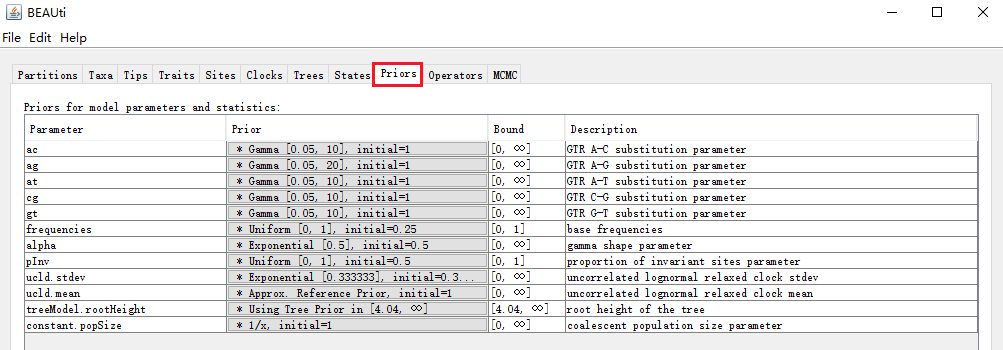 设置参数先验,前提是你知道这些先验信息,否则选择默认就行。
设置参数先验,前提是你知道这些先验信息,否则选择默认就行。
对于本次示例数据(请注意这句话),我们可以根据jmodeltest的结果对模型参数进行先验设置如下:
(1)碱基转换速率设置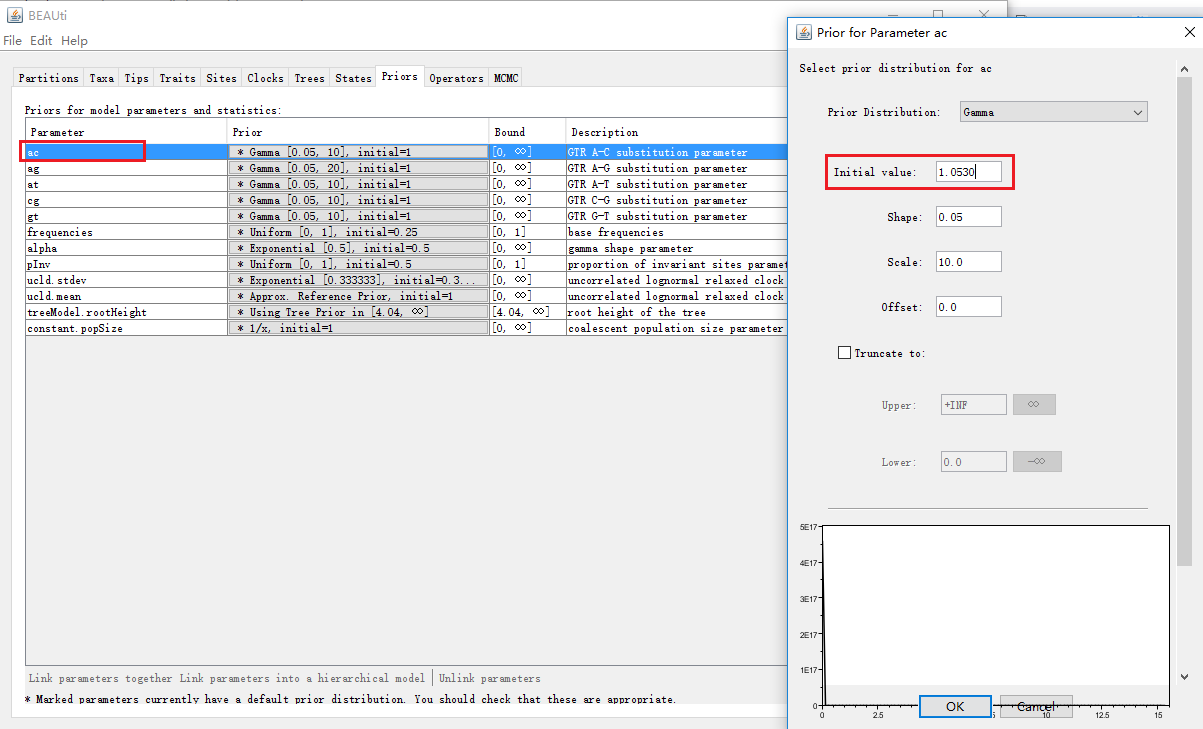
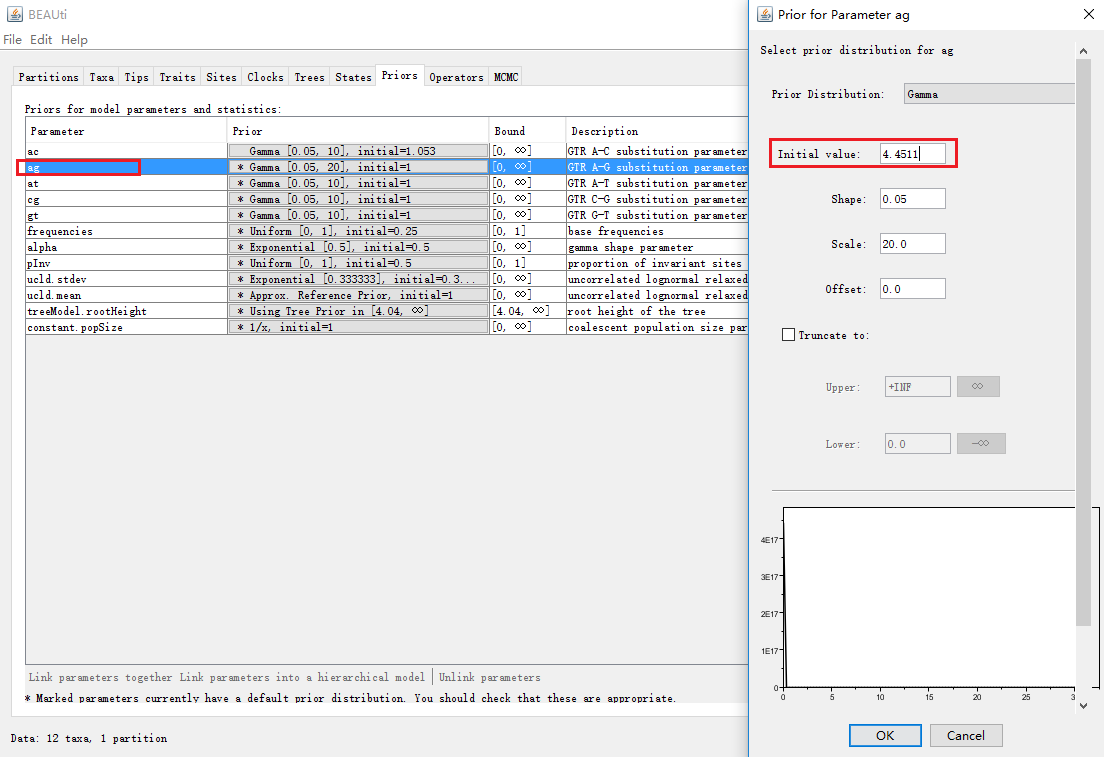
其他的同样操作,这部分的最终结果如下: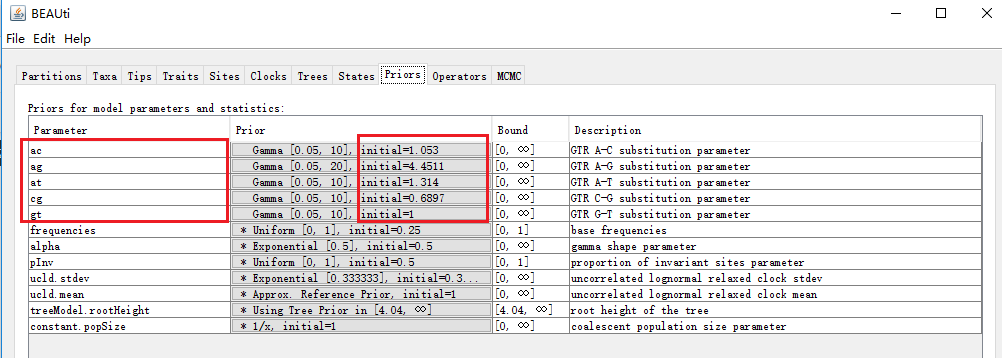
(2)alpha值设置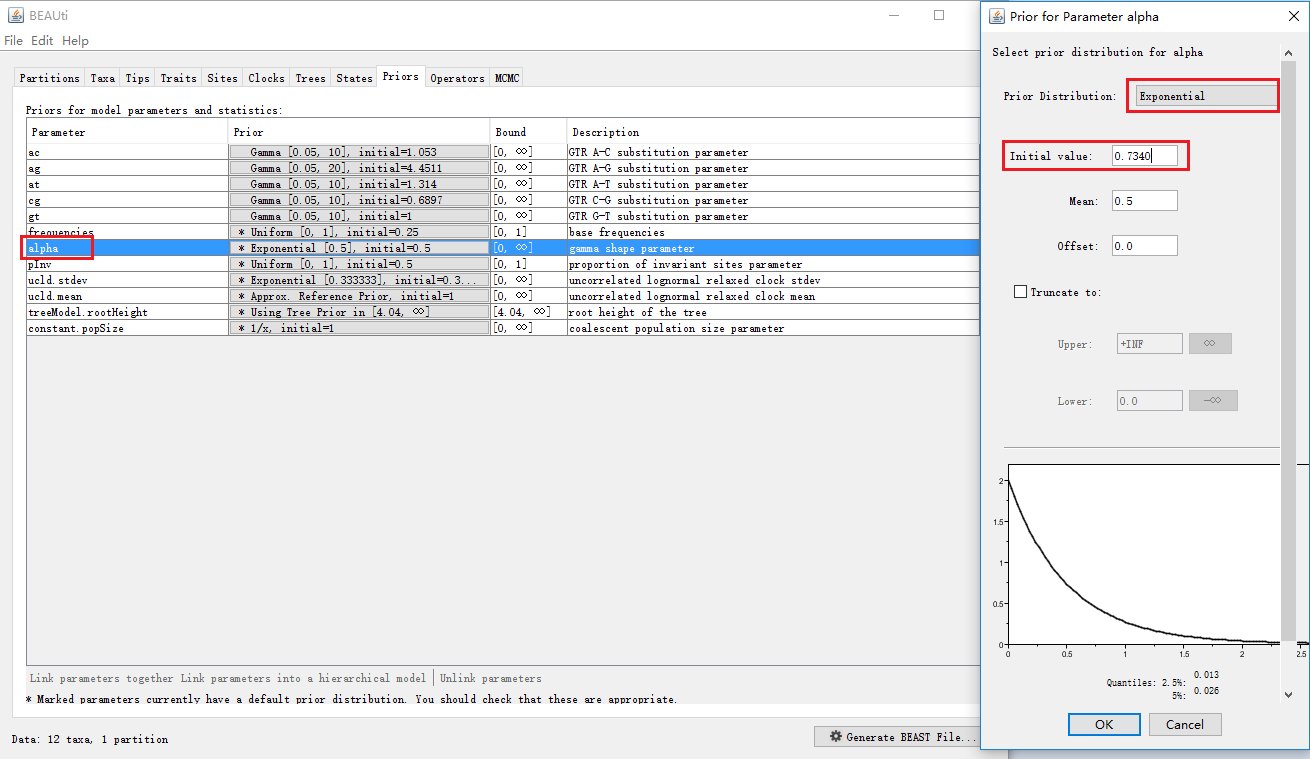
(3)p-inv设置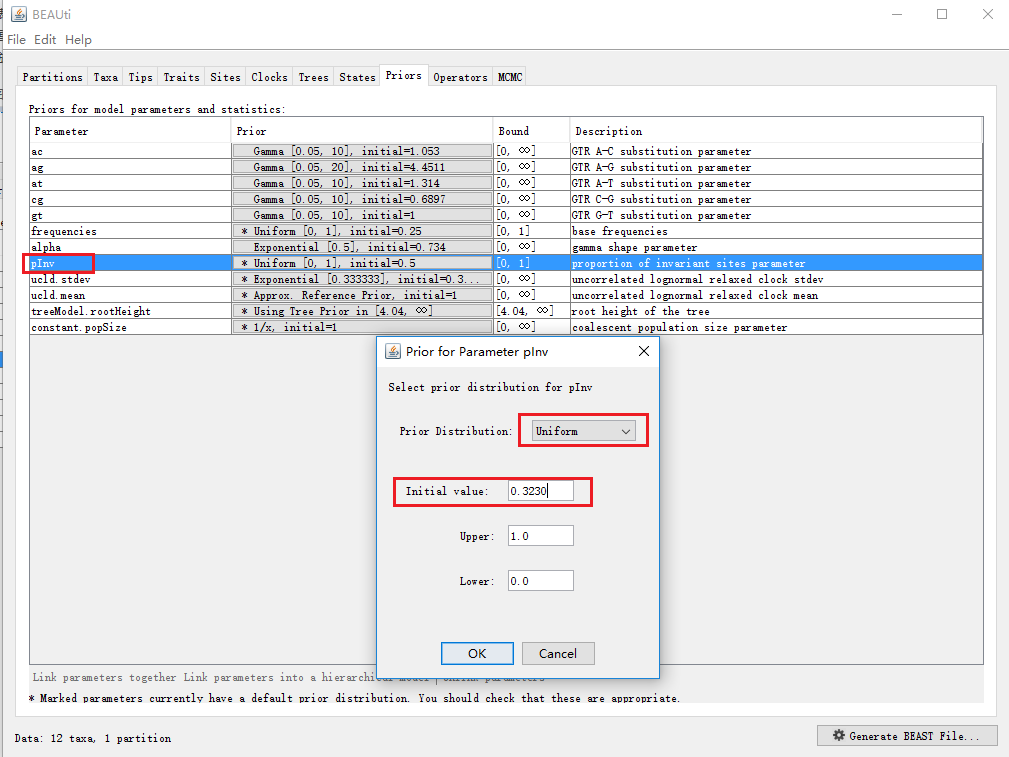
总之,设置任何参数的先验,前提是你有途径或者数据得出。
9. MCMC部分设置
这部分的设置总的来说,因不同数据而异,“总代数=3000 x 序列条数的平方”这个公式仅仅只是粗略地估算(请注意这句话)。
但是,最主要的一点需要保证最后的样本最少得有10,000个。
样本数量=Length of chain(总代数) ÷ Log parameters every(采样频率)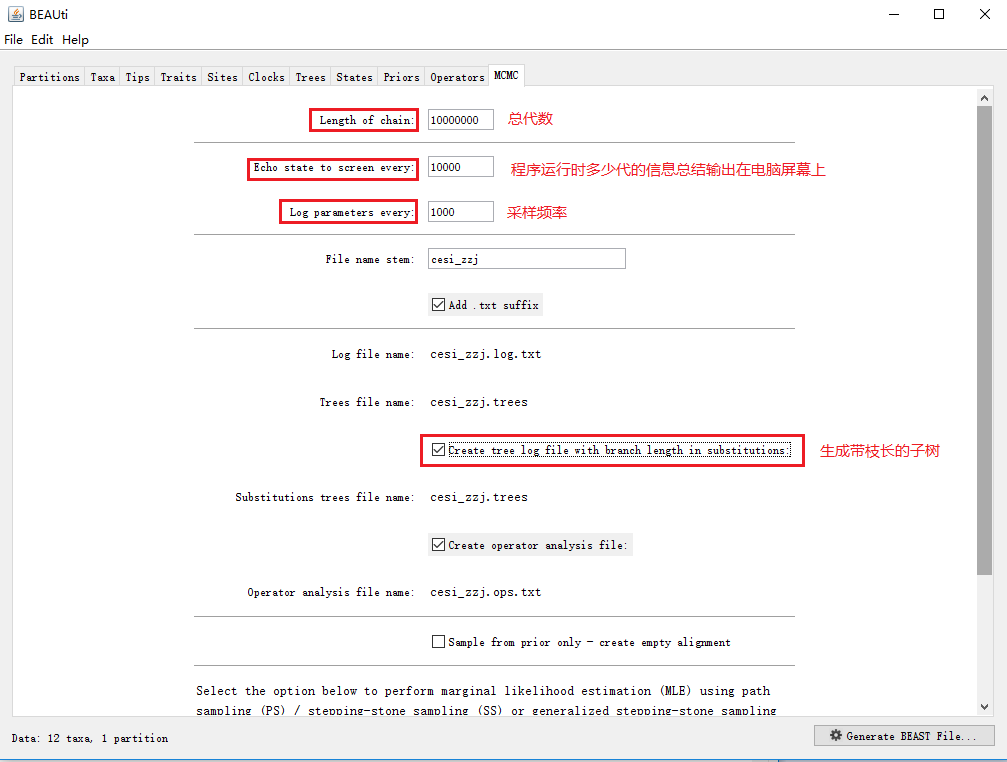
10. 生成XML文件
点击下方红色方框按钮,生成XML文件。
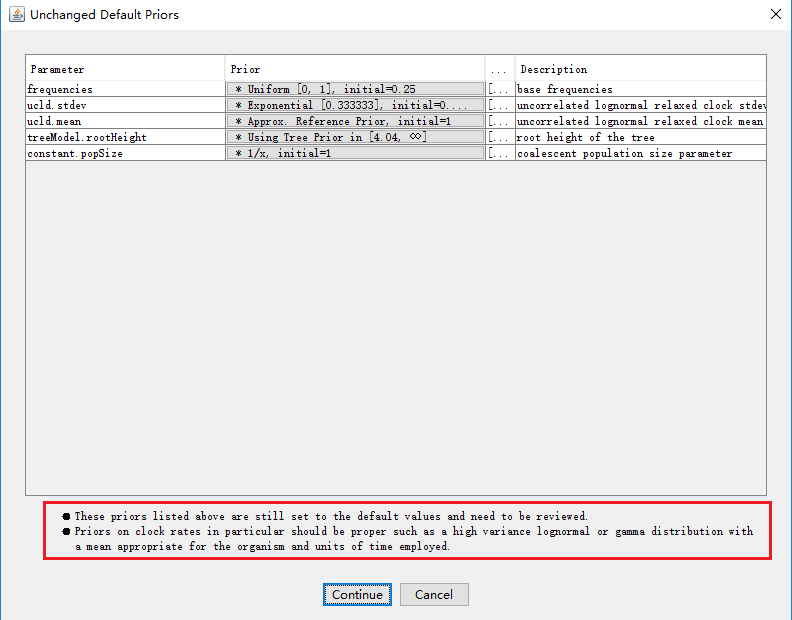
请注意:如果有报错信息,会在上图红色方框处的位置有“!”或者其他颜色字体之类的提示。上图没有错误提示。
11. 运行BEAST
(1)打开上面生成的XML文件,不勾选BEAGLE library,点击运行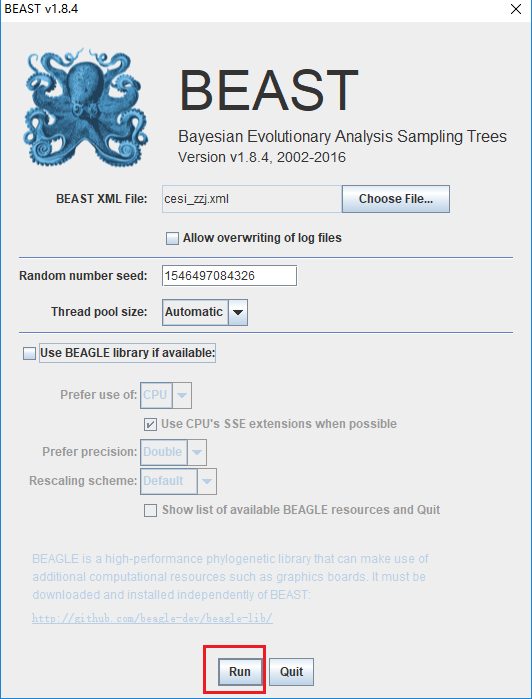
(2)请泡上一杯茶,耐心等待结果,直至所有代数跑完。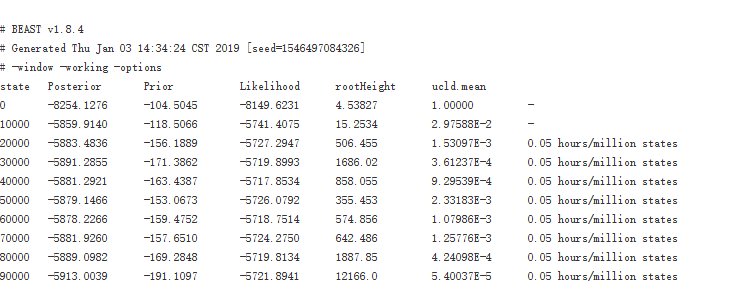
在linux上也可以使用命令行执行beast,代码如下:(进入beast所在目录)
$./beast -beagle_off 1.xml
其中-beagle_off指不使用BEAUGLE
其他参数如下: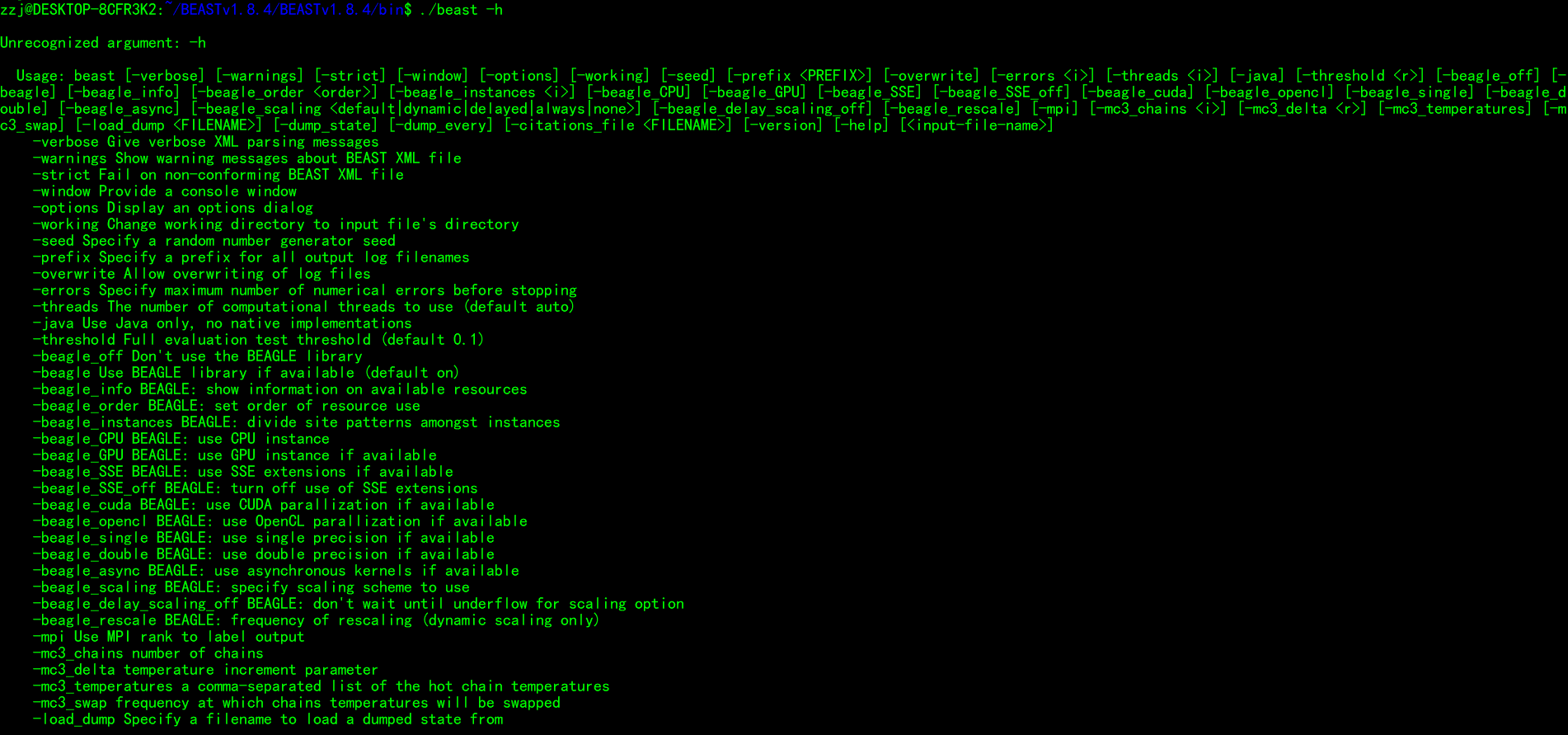
linux上beast怎么顺序运行多个xml文件,用;号隔开每个命令,如下:
$./beast -beagle_off 1.xml;./beast -beagle_off 2.xml;./beast -beagle_off 3.xml
linux上beast怎么同时运行多个xml文件,用|号隔开每个命令,如下:
$./beast -beagle_off 1.xml|./beast -beagle_off 2.xml|./beast -beagle_off 3.xml
同时运行这种方式不推荐,建议顺序运行。
12. 参看参数是否收敛
这个请参考前面部分,不再重复。
13. 获取树
这个请参考前面部分,不再重复。
14. 树的查看及美化
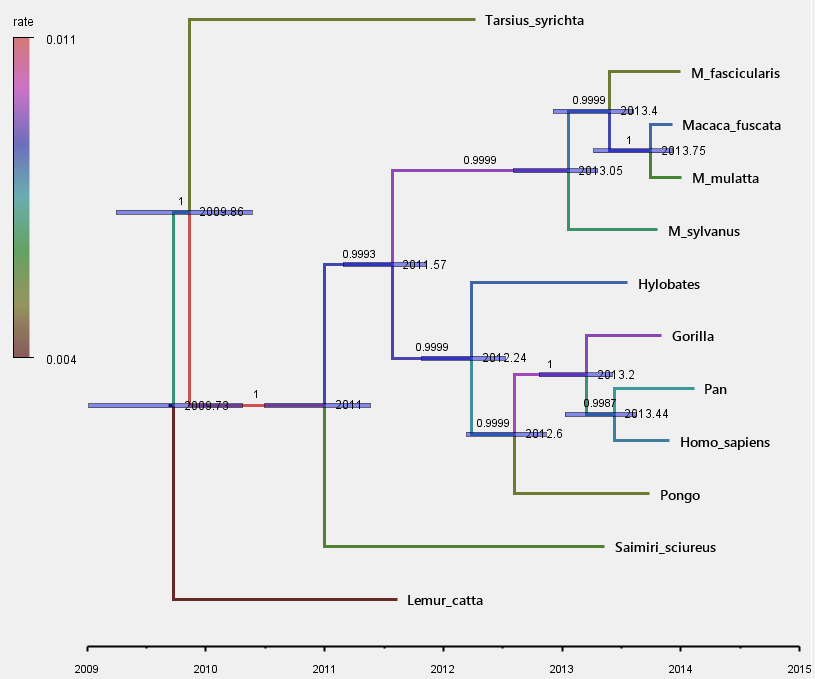
15. 特别注意
BEAST分析过程中,断电了怎么办?
BEAST 1.x 系列没有断点续行功能,可以更名后重新运行,最后合并前后运行的数据;BEAST 2.x 系列有断点续行功能,可以追加运行,有点类似于Mrbayes里的Checkpoint功能。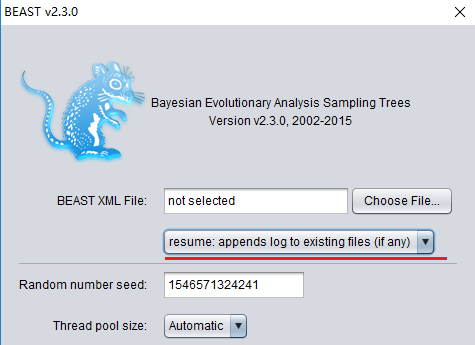
三. 运行XML文件时常见的报错
1. Tree likelihood is zero
起始树的似然为 0 或小于可以使用双精度浮点数表示的最小正数。
这种情况一般当序列比较多,比如超过100条时容易发生。一般是某个或者某些参数的取值范围设置不当引起,比如起始树不符合某些 tMRCA 先验的最大值或最小值,也会发生这种情况。这种情况下,把 tMRCA 先验或者进化速率先验的取值范围调宽点就行。
另外,可以通过命令行的方式打开和运行beast(代替鼠标双击),这样在程序出错时崩溃时会在终端显示详细的报错信息,也能帮助我们排错。
2. 提示java内存不足或者溢出
当数据量多导致计算量非常大时,会出现这种情况。
① 对于windows系统,可以先打开“命令提示符”,以更大的Java内存来运行beast,示例如下:
java -Xms1024m -Xmx1024m -jar lib\beast.jar
可以把调用的最小内存(Xms)和最大内存(Xmx)都调高,示例都调成了1024M,也就是1G,实际中可以给更多的运行内存(取决于你的电脑配置)。
② 对于linux系统,beast这个程序文件其实是可以用文本编译器打开的,打开后,直接在末尾java内存这里的参数进行修改: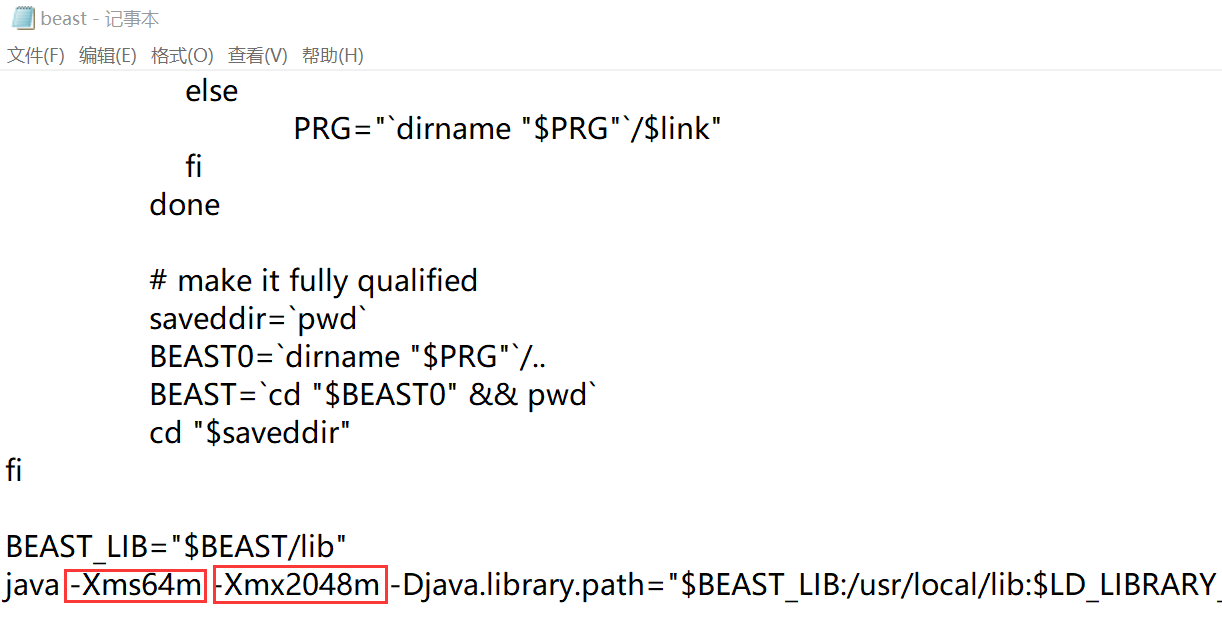
可以发现,软件默认值给的还是非常低的。
对于Linux版本的其他软件,比如TreeAnnotator等,可以以同样的方式调大调用的Java内存。
最后的话:beast这个软件功能非常多,是很多分析的载体,笔者这里写的这些仅仅是入门的冰山一角,加上Beast 1.x系类和Beast 2.x之间不是简单的升级关系,谁也替代不了谁,需要在实践中不断学习这个软化,并反复读它们的相关原版说明书!
转载请注明出处。如有错误,欢迎及时与我反馈,一起交流学习。
2019年1月3日初稿
最近修改时间:2021年8月30日
最后的最后:
打个广告,请关注微信公众号:生信雀。
立个flage:如果微信公众号超过5000人关注,就去B站开个视频教程,Up主。

若有收获,就点个赞吧
0 人点赞
