备注:本文档续“Python进行常见序列处理(1)”,因为写在一个文档里每次编辑有点卡。
十五. 解决Geneious无法完整加载序列名称
Geneious是生物学数据分析一款非常强大的软件,该软件是付费的,该软件的具体功能这里不做介绍。
如果序列名称是我们自命名的,比如是“MG812378|Sparrow|USA-Minnesota|201710”这种“GenID+ 宿主+采集地区+采集年月”的格式,序列导入 Geneious软件后,软件可能无法识别全我们的自命名序列(仅仅是可能),比如只识别了前6个字符,这样可能会出现一些名称重复的序列。
我们可以采取在每条序列名称前加上数字,来解决这个问题。
附代码如下:
seq=open(r"h:\seq.fasta") #需要处理的序列,对于每条序列,要求格式为:每条序列,其名称占一行,序列占一行的fasta格式out=open(r"h:\seq-out.fasta","w")a=[]for line in seq:a.append(line)for n in range(len(a)):s=a[n]if n % 2 == 0:k=n/2 + 1s2=s.replace(">","")s2=s2.replace(" ","_") #将空格换成_,不需要可将此行注释out.write(">"+str(k)+ s2)else:out.write(s)seq.close()out.close()print "OK"
十六. 提取序列的宿主、采集地点、采集时间信息
虽然一直在写代码,但是最近比较忙,没有时间整理,趁今天周末,放点福利(硬货)。<br /> 最近从一个病毒**核苷酸gb文件**里提取信息的时候,顺便写了个脚本,可以提取宿主、采样时间和采样地点信息,感觉这个需求还是很大的。当然了,目前已经有些软件可以很好地实现这个功能,比如BioAider、Phylosuite,笔者也一直在用它们提取gb文件信息,非常方便。这里的python脚本仅供学习交流,相关算法也需要优化。<br />
1. 准备文件
(1)用于提取信息的核苷酸gb文件(氨基酸gb文件笔者没有测试过…)
示例数据:sequence-test.zip
(2)Python3
2. 代码实现
f1=open(r"C:\Users\j\Desktop\sequence-test.gb") #核苷酸gb文件路径out=open(r"C:\Users\j\Desktop\out.txt","w") #输出文件路径date_str=f1.read()a=[]a=date_str.split("//")first_line="GenID"+"\t"+"Host"+"\t"+"Country"+"\t"+"Collection_date"+"\n"out.write(first_line)for n in range(len(a)-1):b = []b = a[n].split("\n")seqID=""host="N/A"country ="N/A"collection_date="N/A"for m in range(len(b)):if b[m].find("LOCUS")!=-1:seqID=b[m][10:22]seqID=seqID.strip(" ")if b[m].find("/host")!=-1:host_str=b[m].strip()host_str= host_str.replace('"','')host = host_str[6:]if b[m].find("/country")!=-1:country_str=b[m].strip()country_str= country_str.replace('"','')country = country_str[9:]if b[m].find("/collection_date")!=-1:collection_date_str=b[m].strip()collection_date_str= collection_date_str.replace('"','')collection_date = collection_date_str[17:]S= seqID+"\t"+ host+"\t"+country+"\t"+collection_dateout.write(S+"\n")print("Fished")f1.close()out.close()
3.提取效果
打开生成的out.txt,将内容全选后复制到Excle表格里(不要问我为什么不直接生成.csv格式的表格文件,懒….),
示例数据效果如下: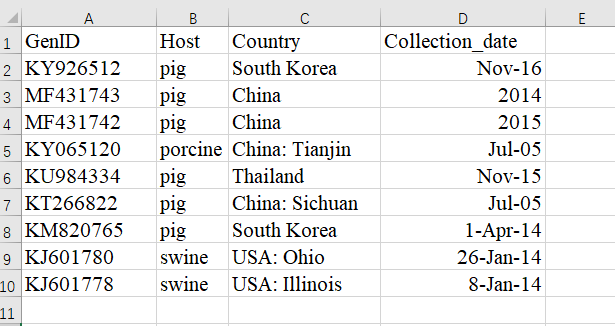
十七. 利用“索引寻找”关联信息并顺序输出
该功能是前面第九节功能的加强版,可以完成第九节的代码的功能,并且应用范围更广。
1.待关联的名称
2.信息多的组合名称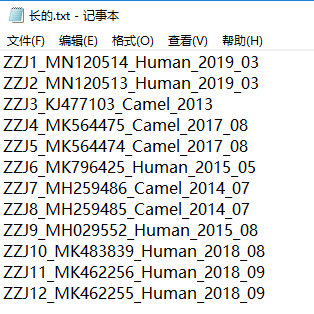
3.实现代码
f1 = open(r"C:\Users\j\Desktop\短的.txt","r") # 短的名称f2 = open(r"C:\Users\j\Desktop\长的.txt","r") # 长的名称out=open(r"C:\Users\j\Desktop\outfile.txt","w") # 输出文件a = []b = []for line1 in f1:line1 = line1.strip()a.append(line1)f1.close()for line2 in f2:line2 = line2.strip(" ")b.append(line2)for i in range(len(a)):flag = 1s = a[i].strip()for n in range(len(b)):chang = b[n].strip()if chang.find(s) != -1:out.write(s+ "\t"+ chang+"\n")flag = 0breakif flag == 1:out.write(a[i].strip()+"\n")print('Finshed')f1.close()f2.close()
4.运行效果
打开输出文件,发现运行结果按照“短的.txt”文件里的名称排序如下:
注意点:笔者示例数据只用了几条,而实际中,我们需要关联信息并排序的序列可能有成千上万条。
十八.制作反向互补序列(序列可以包括简并碱基)
1.序列要求:标准fasta格式序列,如下:
2.实现代码
seq=open(r"C:\Users\j\Desktop\dai.fasta","r")out=open(r"C:\Users\j\Desktop\dai-out.fasta","w")a=[]for line in seq:line=line.strip()a.append(line)for i in range (len(a)):if i % 2==0:out.write(a[i]+"\n")if i % 2==1:a[i]=a[i].translate(str.maketrans('ACGTacgtRYMKrymkVBHDvbhd', 'TGCAtgcaYRKMyrkmBVDHbvdh'))a[i]=a[i][::-1]out.write(a[i] + "\n")
3.效果

若有收获,就点个赞吧
0 人点赞
