**写在前面的话:RaxML是最大似然法(ML法)建树的经典软件,可以建有根树,支持服务器上多线程运行。而且RaxML是目前为数不多支持ML法多基因联合建树的软件,可以说是一款功能非常完备的软件。但是RaxML版本多,参数巨多,让很多初学者望而却步,故将其原版说明书进行翻译,希望大家能熟练掌握这门工具进行系统发育树的构建。**<br />**参考文献:《The RAxML v8.2.X Manual》**
特别补充(写于2021年12月23日):RaxML后面更新换代了,新版本为RaxML-ng,算法有点改动(效仿了IQ-TREE)从而计算速度得到了加快。RaxML-ng最突出了特点是针对核苷酸序列增加了其他很多模型(RaxML里针对核苷酸替换只有GTR模型)。但是请注意,如果对进化模型稍微有了解(包括里面的数学知识)的话,GTR模型是参数最丰富的模型,也就是说就算不测最佳模型,核苷酸直接使用GTR模型也是相对正确的。但是目前论文里掀起的测最佳模型然后建树的热潮(包括笔者也经常测进化模型,然后使用最佳的进化模型来建树),无奈,RaxML-ng只得增加一些奇怪的模型。但如果您真的去钻研了进化模型和里面的数学知识,您就能理解上述笔者所言,然后再决定要不要反驳上述笔者的观点。
尽管RaxML更新了,但“万变不离其宗,一叶而知秋”。参数对应的生物学含义基本七七八八差不多,这份教程写于RaxML更新前,供大家参考。
一.线程问题
- 线程固定在CPU上?
如果可以,将同一线程固定在同一CPU上,即不让同一线程在不同CPU之间切换,可以提高处理速度,固定线程在CPU上的方法:将axml.c 文件里的define _PORTABLE_PTHREADS注释掉。
但是我不推荐将线程固定在CPU上。
- 线程的选择问题
①小于500个位点的DNA序列,使用单线程就可以,也就是使用单线程版本的RaxML就可以。
②单个基因在1000个位点左右的DNA序列,使用2到4个线程就可以,更多位点就需要更多线程,可以自己权衡。
③超过1000个氨基酸序列位点的基因,想要有效运行,至少需要16线程。
④如果变异速率模型是GAMMA模型,它比CAT模型需要更多的线程(CTA需要的线程只要GAMMA的四分之一),少于50条序列不建议使用CAT变异速率模型,CAT被用于加速具有许多分类群的大型数据集的计算(原文)。
警告,不要使用基于CAT的似然值来计算树的拓扑结构,你可能会得到一个错误的树(原文)。
二.检查序列格式
RaxML对序列格式和序列内容要求非常严格,建树前先检查序列格式是否错误。
1. 序列的名称禁止出现空格,制表符,换行符,:,()或者[ ]。
2. 不能有相同名称的序列。这个可能是我们将其他格式的序列变成phylip格式的时候,只保留序列名称前8个或者10个字符造成的。
3. 不能出现不同名称但序列一样的这种序列。
4.序列不能完全由未知符号组成,即序列里不能全是这样的字符:如氨基酸序列里不能全是X、?、*;如DNA序列里不能全是N、O、X、?。(红字部分)
注意:①RaxML会自动移除你序列文件里的相同序列和不确定的序列。
②如果是一个分区模型的分析,则相应的模型文件modelFileName.reduced也会被读写。 RAxML遇到相同的序列名称或未确定的序列或非法字符,它将退出并报错。(②这种情况是多基因联合建树)
三. RaxML参数
1.使用前可在命令终端输入raxmlHPC –h查看该软件的帮助文档,可以在帮助文档前一部分看到以下内容。(实际上会出现非常多的内容,包括对每个参数的解释)
(1)linux系统命令端输入raxmlHPC –h 后第一行可能出现下面这样的字符,
raxmlHPC[-SSE3|-AVX|-PTHREADS|-PTHREADS-SSE3|-PTHREADS-AVX|-HYBRID|-HYBRID-SSE3|HYBRID-AVX]
显示该软件可以进行编译的RaxML版本,不同版本使用符号“|”隔开。
版本按原理分类:
① MPI版本 (message passing interface) ,处理大的运算之后的数据,比如进行100次或者1000次自举。
② PThreads(multi-core shared memory systems),多线程版本。应用于非常长的序列分析,电脑必须是多核或者说是多线程的。
③ HYBRID版本,联合了MPI版本和PThreads版本,先用PThreads去计算单一树的似然值,再用MPI去处理自举检验或者对不同的共享的储存节点进行独立树的搜寻。
【参数介绍】
(2) -s sequenceFileName -n outputFileName -m substitutionModel
这一行的参数比较简单,就是输入-s phy格式序列文件名字 **-n 输出文件名字 –m 核苷酸或氨基酸替代模型+变异速率模型**
{注意:替代模型和变异速率模型,就是你用模型寻找软件得到的最佳模型,比如:GTRGAMMAI(即GTR+G+I),其中GAMMA是变异速率模型即G。}
方括号中为可选项。
(3)-a 设定每个位点的权重,必须在同一文件夹中给出相应位点的权重,比如下面这样的:
5 1 1 2 1 1 1 1 1 1 1 2 1 1 3 1 1 1 1 1 1 1 1 1 1
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 4 1 1 1 4 1 1
-a 后面接包含权重的文件的文件名。该文件必须和你用来建树的那个序列文件在同一个文件夹内。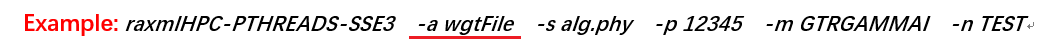 (其中wgtFile是包含权重的文件的文件名,后面的参数暂时先别管,后面会一一讲到。这个例子用的是PTHREADS-SSE3版本的raxml)
(其中wgtFile是包含权重的文件的文件名,后面的参数暂时先别管,后面会一一讲到。这个例子用的是PTHREADS-SSE3版本的raxml)
除非特殊,一般情况下,我们不用-a这个选项,也就是不输入 -a wgtFile
(4) -A 指定一个二级结构替换模型,可以使用的模型有:S6A, S6B, S6C, S6D, S6E, S7A, S7B, S7C, S7D, S7E, S7F, S16, S16A。默认的模型是S16,即16 state GTR model
你如果想指定一个特殊的二级结构替换模型,要使用-S来指定这个包含二级结构的文件
说明:每个参数在代码里的相对位置是可以随意的。除非特殊,一般情况下我们也不输入 –A。
(5)-b 设定bootstrap起始随机数,默认是关闭的。这个选项可以让你开始非参数bootstrap,允许重复运行。比如我们可以输入12345或者1234756去制定这个随机数等等。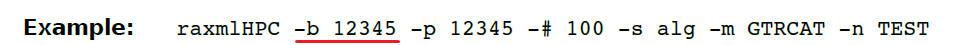
但是,如果你使用的是并行版本raxmlHPC-MPI,指定了随机数也不能重复运行。
(6)-B 用于切断基于MR的自举停止标准,对大多数一致树的收敛有积极作用,默认值是0.03。可调节范围是0.0~1.0。
一般情况下,我们用默认值就行,默认值0.03是基于经验得出来的,在实际中不需要输入-B。
(7)-c 当使用CAT变异速率模型时去指定_distinct rate categories,默认值是25。
注意,如果你使用的是Gamma变异速率模型,不管你将-c 设置为多少值都是没有作用的,因为_Gamma只有4个变异速率。
(8)-C 激活“-L”和“-f i”选项的冗长输出,默认是关闭。
(9)-d 完全随机的搜索进化树,而不是从maximum parsimony tree 开始。在100 至200 个分类单元间,
该选项可能会生成拓扑结构完全不同的局部最大似然树。默认值是关闭。
(10)-D ML法搜寻收敛的标准。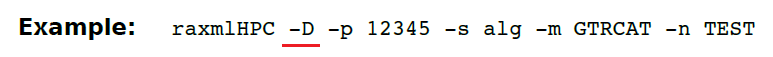
在非常非常大的数据集或许可以采用这个,但是可能找到的是不好的树,所以这个选项默认在关闭的。
(11)-e 对最后最佳的树拓扑结构在似然值对数上设置模型最佳的精确度。默认值0.1。
默认值:0.1 表示对于模型不使用估算的不变位点的比例。
- 表示对于模型使用估算的不变位点的比例。
这个允许你去指定似然差异的上限,当你在RaxML上使用GAMMA变异速率模型或使用-f e 选项评估一棵树或使用-f n 选项及相似选择去评估树的枝长,对-e 进行设置会让你的模型参数最佳化。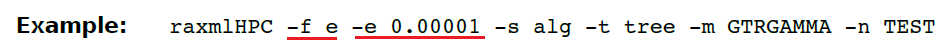
默认值0.1似然值对数适用于大多数数据的处理。大多数情况下我们不用输入-e。
(12)-E 指定一个位点排除文件的文件名,即某些位点不用来建树。在这个位点排除文件里,你需要把想要排除的位点进行详细说明,制作一个类似.nex格式的文件。
在多基因联合建树的时候,你通过-q 选项指定了一个模型分区的文件,当你使用-E进行位点排除时,模型分区文件里指定的分区信息也会相应自动更新。说白了就是程序会先处理-E选项,再处理-s选项或-q选项。
比如我要排除100-199位点(会包括第100和第199)和200-299位点,一个简单的排除位点文件里包含下面两行:
如果不需要去排除位点我们不需要输入-E,同样的,如果不需要多基因联合建树,也不需要输入-q。
(13)-f 选择算法,后面接各种算法:a|A|b|B|c|C|d|D|e|E|F|g|G|h|H|i|I|j|J|k|m|n|N|o|p|q|r|R|s|S|t|T|u|v|V|w|W|x|y
现在一一讲解:
(13.1)-f a 快速自举分析(rapid Bootstrap analysis),可以快速进行自举分析找到一棵最大似然树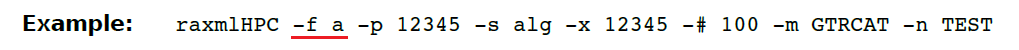
(13.2)-f A 对你提供的有根参考树(通过-t选项)进行边缘祖先的估算。
结果会再次输出一棵有根二叉树,但是内部节点的标签在树上的对应位置用于联合祖先序列。它会生成两个文件,一个是包含了每个内部节点的可能边缘,另外一个文件包含了对真实祖先序列的推测。
(13.3)-f **b ** 基于多样树,比如来自自举的多样树(通过-z选项加载),来对已知的最佳的ML树(通过-t 选项加载)画出二分信息。
-f b 这个选项相当于是用ML法建树后,再对树的相关信息进行进一步探索。
(13.4)-f **B **优化枝长标尺和其他模型参数。

通过-t 选项输入一棵带有枝长信息的树, 枝长本身不会被最佳化,只是最佳化枝长标尺。
多基因联合建树时,认为所有的分区都有相同的枝长比例。通过-q 包含多个模型信息的文件。
这一步操作相当于是在建树后对标尺进行优化,尤其是多基因联合建的树。
(13.5)-f c 核对比对后的序列是否可以被RaxML读取。
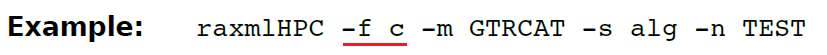
(13.6)-f C 祖先序列检测。
输入一颗已知的最好的ML树,再输入一组假定祖先候选序列。
(13.7)-f d 快速hill-climbing算法,默认值开启,不需要设置。
(13.8)-f D 执行一个或者多个快速hill-climbing算法。当产生的树比较多,计算机没有资源或者时间进行bootstrap replicates时,建议开启这个选项。

(13.9)-f e 为给定的已知树优化模型参数+分支长度(需要通过-t 选项输入一棵树)
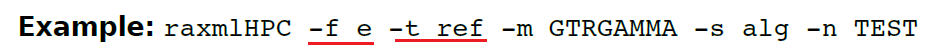
图中ref是树文件的名字。
(13.10)-f E 执行非常快速的树搜寻试验,这个选项,目前仅仅用于测试。
(13.11)-f F 执行非常快速的树搜寻试验,这个选项,目前仅仅用于测试。
(13.12)-f g 比较一棵或者多棵树在每个位点的似然值,通过代码-z 输入树文件,生成可以被CONSEL显示的饼状图格式文件。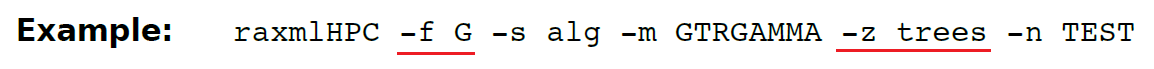
模型参数只能预估树文件里第一棵树。想要将模型参数评估每棵树使用 **-f G 。**
(13.13)-f h 在最好的树和其他树之间进行似然值对数检验。(SH-test)。-t 导入最好的树,-z 导入其他一堆树。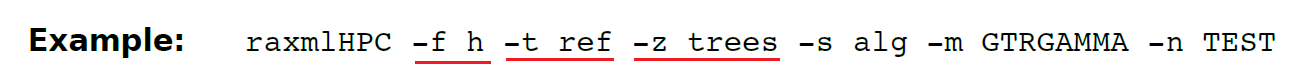
-模型参数只能预估-z导入的树文件里第一棵树。想要将模型参数评估每棵树使用 **-f H(大写) 。**
(13.14)-f i 在一棵参考树(a reference tree)和多个树之间((e.g., from a bootstrap))计算IC和TC分数。-t 导入参考树,-z 导入其他多个树。
默认的TC / IC计算并不完全按照MBE中的描述进行。 要让RAxML完全按照本文所述进行操作,请编辑文件bipartitionList.c,通过注释掉或删除下面这行的记录。
#define BIP_FILTER
(13.15)-f I 一个简单的树生根算法,用于无根树。即把无根树变成有根树。- t 导入无根树文件。

(13.16)-f j 从原始对齐序列文件生成一堆引导的对齐序列文件。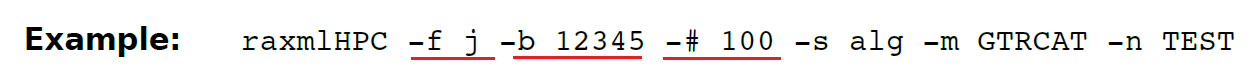
通过-#设置自举值。如果要为分区分析的数据集生成那些BS复制对齐,则还需要通过-q在此处指定分区文件,因为RAxML基于每个分区重新对样本进行重新采样!(多基因联合建树)
(13.17)-f J 计算一棵树的SHlike支持值。
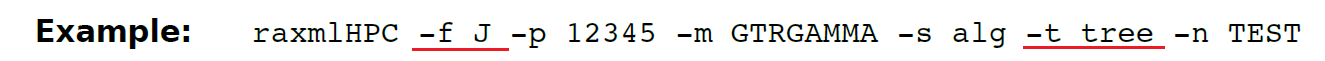
(13.18)-f k 使用枝长窃取算法修复有缺失数据的分区数据集中的长分支长度,此选项尝试修复缺少数据的分区数据集中的非常长的分支长度问题。同时还需要另外三个参数:“-t“”-M“,”-q“。
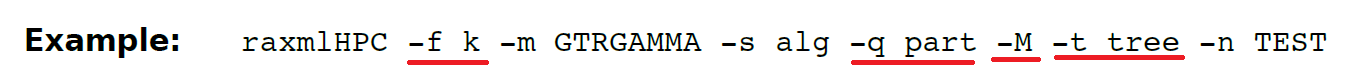
它将输出具有较短分支长度的树,但具有相同的分支似然得分。输出结果在一个名称为RaxML_stolenBranchLengths.TEST的新树文件里。
(13.19) –f m 比较两大类树的二分法。输出结果在一个RAxML_bipartitionFrequencies.outpuFileName文件里,文件里包含两个集合的pair-wise bipartition frequencies 。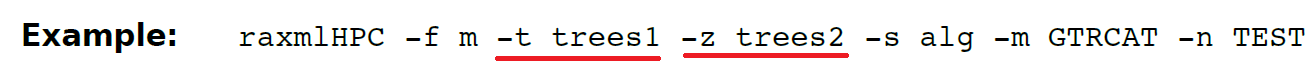
注:每个树文件里有多棵树。
(13.20)-f n 计算树文件中包含的所有树的对数似然得分,通过-z 导入树文件。
模型参数只能预估树文件里第一棵树。想要将模型参数评估每棵树使用 **–f N(大写)。**
(13.21)-f o 使用此选项,通常会获得稍好的似然得分,而运行时间预计会增加2到3倍。
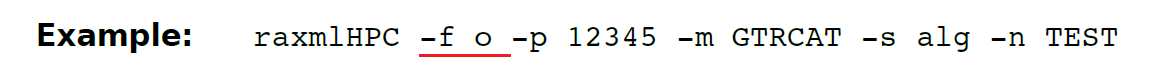
(13.22)-f p 执行纯粹的逐步MP法(最大简约法)添加新序列到一棵不完整的起始树。
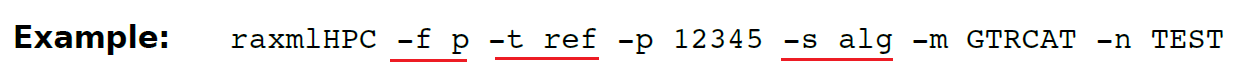
(13.23) –f q 即 _fast quartet calculator,极少用到,省略。_
_(13.24)-f r 计算一个树文件里所有树的成对碱基的_RobinsonFoulds(RF)距离。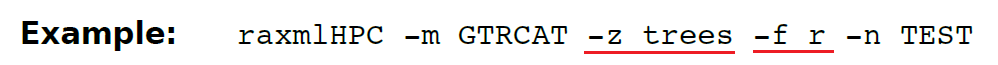
(13.25)-f R 比较一棵大的参考树和许多小树之间的_成对碱基的_RobinsonFoulds(RF)距离。
(13.26) -f s 将一个多基因分区的序列集变成独自的子序列集。子序列集的序列名字将根据你分区的命名来命名,会分别产生每个分区的.phy文件。(也就是将联合后的多基因再分开建树。这波操作可以)
(13.27)-f S 由进化放置算法得到的测试灵感计算特定位点的放置偏差。平均位点放置距离相对较低的区域,意味着这个区域在其整个长度上有相对稳定的系统发育信号。
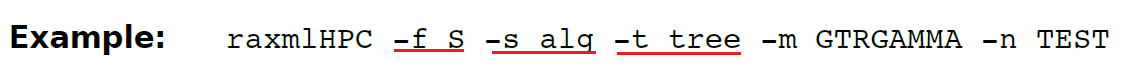
-s 导入的是一组对齐序列的某个区域片段,-t 导入参考树,一般是已知的最佳ML树。
输出结果在一个RaxML_SiteSpecificPlacementBias.TES的文件里,可以使用比如gnuplot这样的描点工具得到图形。
图形示例: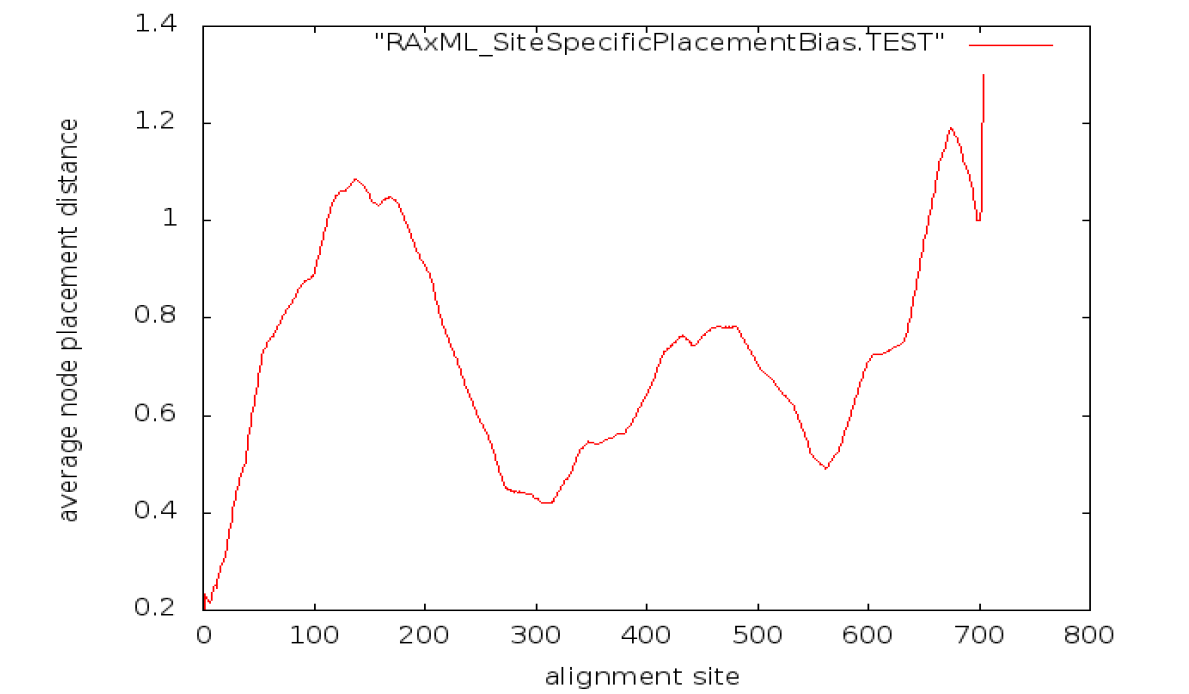
这个图形的意思就是,比如用比对后的全基因组序列建好ML树后,再用这个比对后的全基因组序列对进行-f S ,查看该全基因组序列哪个片段的平均节点放置距离相对较低,则该片段有相对稳定的系统发育信号。 这个很有意思!
(13.28)-f t do randomized tree searches on one fixed starting tree。
此命令将执行指定数量(通过N或#)的随机树。与标准搜索算法的不同之处在于,给定一个起始树,它将以随机顺序应用SPR(子树修剪重新移植)移动,从而可以使更好的树变得更适合。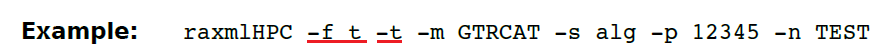
- t 选项表示使用随机树。
(13.29)-f T 在独立模式下通过快速自举搜索对ML树进行最终彻底优化. 此选项允许进行更彻底的树搜索,使用较少的延迟,即更多在独立模式下,详尽的SPR移动。 该算法通常在通过-f a 完成的搜索的每一端执行。
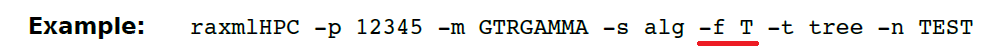
这个选项用得比较少。
(13.30)-f u ,-f v , -f V很少用到。
(13.31)-f w 在一堆树上进行ELW测试。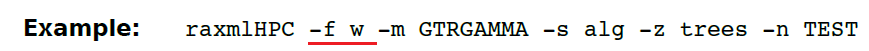
模型参数只能预估树文件里第一棵树。想要将模型参数评估每棵树使用-f W(大写)
(13.32)-f x 计算成对ML距离,ML模型参数将在MP起始树或通过-t 输入的自定义树进行估计,仅允许基于GAMMA的变异速率模型。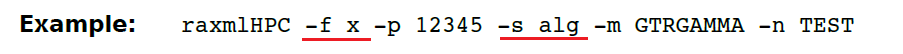
(13.33)-f y 使用简约法将一堆环境序列分类到参考树中,您需要使用非全面的参考树和包含所有序列的对齐来启动RAxML(引用+查询)

(14) –F 为非常大的树启用CAT模型搜索ML树,最后不切换到GAMMA模型(节省内存)。如果你想避免因为时间或内存限制(GAMMA需要CAT四倍的内存),你可以使用这个选项。此选项也可以与GAMMA模型一起使用,以避免最终彻底优化最佳得分ML树。该选项默认是关闭的。
注意:可以使用CAT搜寻ML树,但是不要用CAT计算拓扑结构,前文已经提到。
(15)- g 预先分组的名称
(16)- G 通过指定阈值(在ML下使用彻底插入来评估的插入分支的分数)来启用基于ML的进化放置算法启发式算法。此选项可与-f v和-f V选项一起使用,以加快短读取的进化位置。 设置为0.1(10%的分支被认为是插入次数)会产生良好的准确性/速度权衡。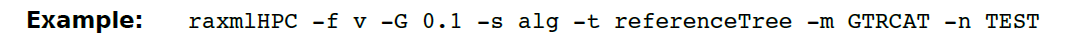
(17)-h 帮助文档。
(18)-H 禁用压缩模式。默认该选项开启,不用管。
(19)- **i **为子序列用于拓扑结构而初始重排设置。程序会自动运行,不需要调节。子树将被插入到距其修剪位置1到10个节点之间的所有分支中。
(20)-I (大写的i) 此选项允许执行bootstrap收敛测试,确定是否已计算足够的BS重复以获得稳定支持值的测试,一个后验,有点类似于BI法建树。在已经计算过之后,例如,100个bootstrap重复,在这里你最初会运行50个重复并检查它们是否足够,然后再运行50个并评估你现在拥有的100棵树是否足够。在超级计算机上进行大规模树搜索时,此选项特别有用。超过1000个序列使用 autoMRE_IGN节省时间。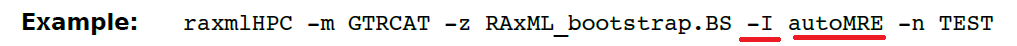
(21)- j 默认关闭。
(22)-J 不用理会。
(23) -k 指定bootstrapped树应该输出分支长度。默认值关闭。
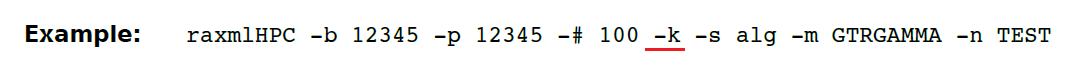
(24)-K(大写) 在RaxML中指定multistate substitution models中的一个(最多30个),可用的模型有ORDERED, MK, GTR。默认的是GTR model。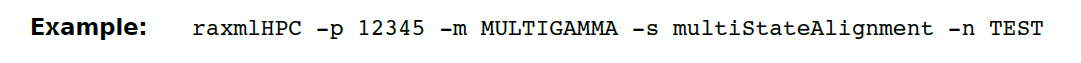
如果有几个分区,其中包含多态字符,那么通过-K指定的模型将应用于所有模型。因此不可能分配不同的模型到不同的多状态分区。(见于多基因联合建树)
(25)-L 计算基于IC的树一致性和总TC值(Salichos和Rokas提出的),即序列相似性阈值。
默认的TC/IC计算没有完全按照MBE中描述的那样进行。要让RAxML完全按照论文中描述的那样做,请编辑文件bipartitionList。通过注释或删除下面行:
#define BIP_FILTER
(26)-m 模型设定(重点)。二进制模型,核苷酸替代模型,Multi-State模型,氨基酸替代模型。
注意:对于分区数据,替代模型可以因分区不同而不同,但是变异速率模型对于不同分区都是一样的(RaxML软件是这样),不能一个分区用CAT变异速率模型,而另一分区在GAMMA下分析。(多基因联合建树),可以通过-m对分区数据导入共同的变异速率模型。
【特别注意】
①和其他软件相比,软件RaxML针对核苷酸的替代模型只有GTR一种。
因此,GTR(核苷酸替代模型)+变异速率模型的组合有以下几种:
-m GTRCAT: GTR approximation
-m GTRCATI: Same as GTRCAT, but with estimate of proportion of invariable sites
-m GTRMIX: Search a good topology under GTRCAT
-m GTRMIXI: Same as GTRMIX, but with estimate of proportion of invariable sites.
-m GTRGAMMA: General Time Reversible model of nucleotide subistution with the gamma model of rate heterogeneity.
-m GTRGAMMAI: Same as GTRGAMMA, but with estimate of proportion of invariable sites
-m GTRCAT_GAMMA: Inference of the tree with site-specific evolutionary rates. 4 discrete GAMMA rates,
-m GTRCAT GAMMAI: Same as GTRCAT_GAMMA, but with estimate of proportion of invariable sites.
加“I”或不加“I”的区别如:
②可用的AA模型有:DAYHOFF, DCMUT, JTT, MTREV, WAG, RTREV, CPREV, VT, BLOSUM62, MTMAM, LG,
MTART, MTZOA, PMB, HIVB, HIVW, JTTDCMUT, FLU, STMTREV, DUMMY, DUMMY2, AUTO,
LG4M, LG4X, PROTFILE, GTR_UNLINKED, GTR。
注意:当使用LG4X蛋白替代模型时,请记住,这个模型实际上比普通的固定蛋白替代模型有更多的自由参数。因此,对于在LG4X下演进的每个分区,都有6个额外的自由参数(3个用于权值,3个用于速率)
③在模型名字后加上字符“X”可以标出用最大似然法估计的基础频率,不标“X”核苷酸序列默认是经验基础频率。

格式:

示例如下:
对于氨基酸序列,可以在氨基酸模型名字后加上F或X,其中F代表基于经验基础频率,X表示最大似然法估计的基础频率。
格式如下:

示例:
④在模型名字前加上字符“ASC”可以通过似然值进行系统发育偏移修正。ASC_仅仅用于单核苷酸变异的DNA数据。
格式:
示例如下:
此外,RaxML还可以自动寻找模型,类似于其他模型寻找软件。
(27)-M (大写) 进行每个分区分支长度的估计。只有与“-q”一起使用才有效果。(多基因联合建树)。将输出单个分区的分支长度以分隔文件,通过使用各自的分区长度计算分支长度的加权平均值。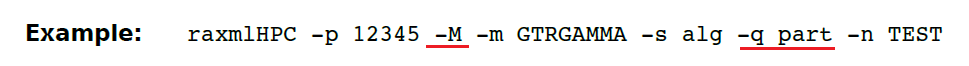
注意:-M 选项默认是关闭的。
(28)-n 指定输出树文件的名字
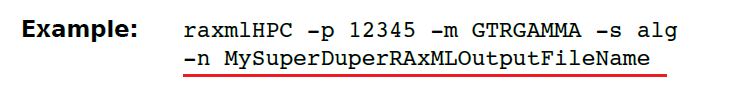
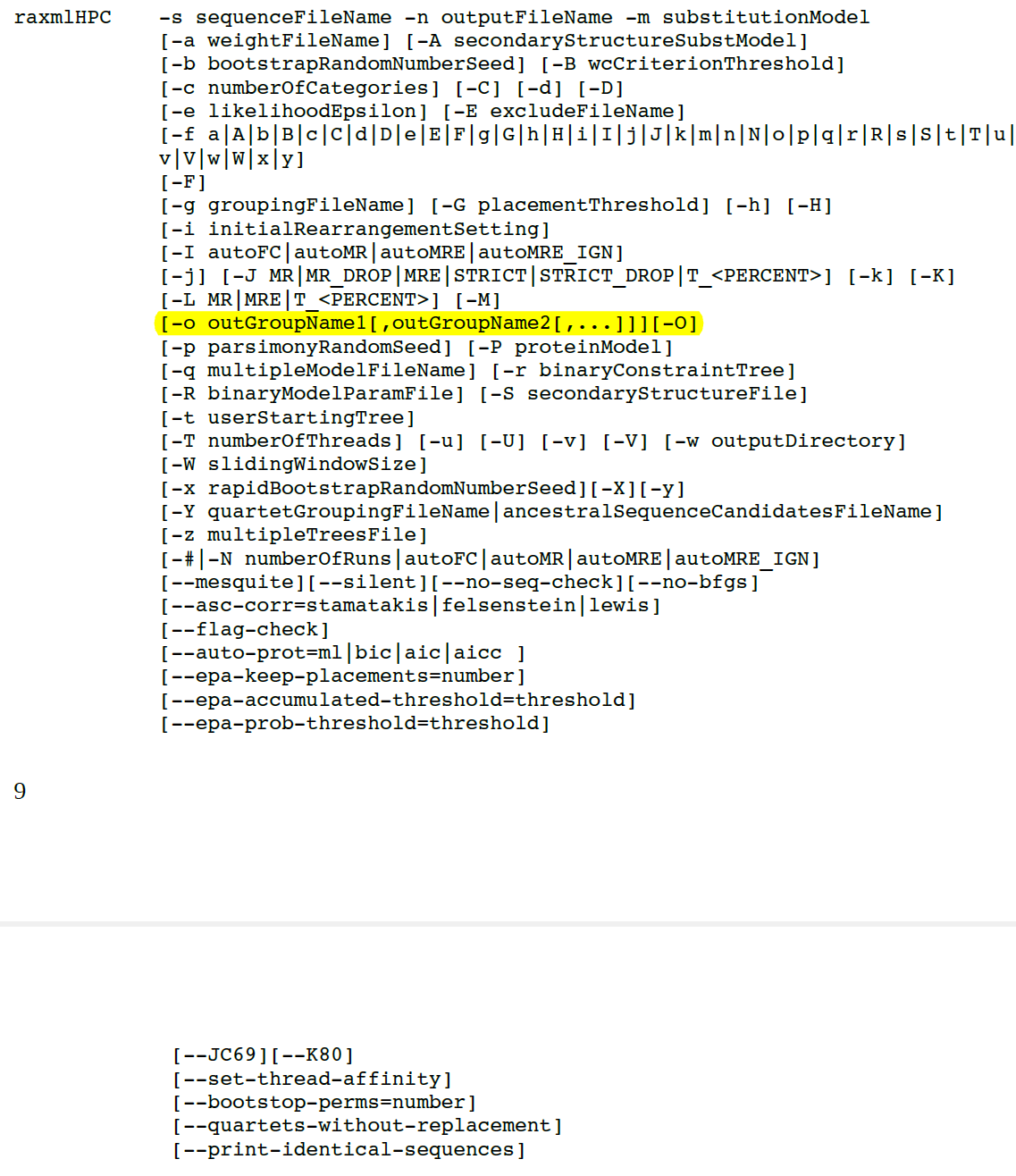
(29)-o 定义外群,用来建有根树。设定外类群如果有两个以上外类群,外群的名称之间不能用空格,而应该用英文的逗号”,”。
重点来了:
①如果设置了多个外群,但这些外群不是单系群(名词了解下),程序会选择外群列表里的第一个作为外群来生根,最终程序只会选择一个外群。
②始终记住外群只是一个画树的选项,这棵树本质上依然是无根树。
来自该程序原文作者的建议:
他通常避免在最初的ML和bootstrap分析里使用外群,而是这么做:
①先只用内群建树,包括ML和BS(bootstrap)过程,相当于先只建无根树。
②建完无根树后,使用EPA (see: http://sysbio.oxfordjournals.org/content/60/3/291.short)去把外群放在树上。
这样做有几个优点:
①你可以使用或测试不同的外群。
②如果你的外群扰乱了你内群的分析,你不用再重新跑一遍分析。
③你可以防止外群影响内群的枝长。
④You get placement probabilities (likelihodo weights) for each outgroup, e.g., how likely
the outgroup is to fall into one branch or another. This also tells you if your outgroup is good
(high probability for being placed into one specific branch) or if it is bad (scatters across the tree)
(30) – O(大写) 关闭核对完全未确定的序列。默认值:check enabled。这个选择我们不用管,不设置。
(31)-p 为简约推断指定一组随机数。可以重写你的结果帮助我能纠正程序。这里我们通常要设置,一般将随机数设置12345就行。
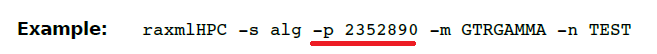
(32)-P(大写) 这个是指定一个用户定义的氨基酸替代模型文件。文件必须包含420行,前400行是一个氨基酸替换速率(必须是一个对称的矩阵),后20行是基于经验的频率。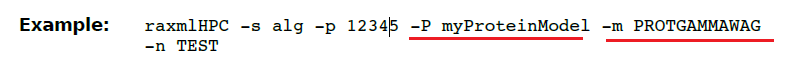
对于上图的例子,如果再同时再用-m添加氨基酸模型,如上图的-m PROTGAMMAWAG ,其中氨基酸替代模型WAG会被忽略,而只会使用GAMMA变异速率模型。
对于分区数据,如果想要用自己的氨基酸替代模型,这个替代模型文件同样是上面的格式去在分区文件里进行定义。(这个自定义时候RaxML本身不包含的一些模型)
(33)-q 指定一个针对分区数据含有多个替换模型的文件,注意以下几点:
①为你比对后的序列的不同区域单独指定替换模型。分区文件也可以包含不同类型的数据,比如你的分区数据里1-500个位数点是核苷酸序列,501-600是氨基酸序列。但是你必须在分区文件里指定数据的类型。
②这对于包含多个基因的序列建树非常有用。
③如果你想使用最大似然法推测基础频率(base frequencies),需要在替代模型的名字后添加“X”,比如DNAX,LGX。
④如何定义分区数据每个分区使用的替换模型。(类似于定义矩阵的列)
- 对于核苷酸DNA序列
(i)在每一行最前面使用字符“DNA”。(因为RaxML只为核苷酸序列提GTR这一种核苷酸替代模型,故写成DNA)
(ii)继续上图,如果多序列比对后的基因1是分散的,怎么定义分区数据?

(iii)为编码位点指定不同的模型。

b.氨基酸数据
(i)为每个分区指定转换矩阵(替换模型)。
注意:RaxML里,氨基酸替换模型的名字必须在每行的最前面,然后再指定每个分区的基因名字。但是对于所有的分区只能指定同一个变异速率模型,通过-m 来指定。
(ii)如果你想对其中一个分区使用你自己的氨基酸模型(即RaxML软件本身不包含的),可以这样。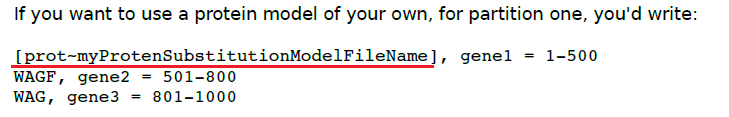
(iii)如果想用最大似然法计算氨基酸数据每个分区的基础频率,可以这样做:(在模型后面加“X”)
c.如果你的分区数据里既有DNA核苷酸序列,也有氨基酸序列,在模型分区文件里可以这样给每个分区定义替换模型。

示例命令如下: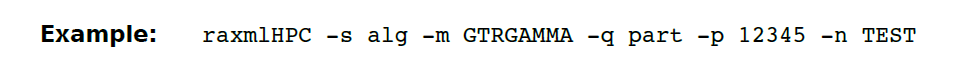
(34)- r 和- R 这两个选项和二进制数据有关,我们用DNA序列和氨基酸序列(AA)建树基本用不到。
(35)- s 指定用于建树的PHYLIP格式文件。
你不用在意序列是交错(interleaved)或顺序(sequential)的排列,也不用管分类单元名称太长(序列名称可以在1-256个字符,需要更长名称可以在文件axml.h去定义 #define nmlngth 256 in file)。
但是你需要注意的是,在分类单元名称和序列之间总是需要有一个空格。
RAxML现在还可以解析FASTA格式。 如果RAxML注意到它无法解析phylip格式,它将尝试将对齐文件解析为FASTA格式。 到目前为止,它已经能够解析所有FASTA文件。
(36)-S(大写)指定一个二级结构文件。具体格式参考原文说明书。
Specifying secondary structure models for an RNA alignment works slightly differently than passing other data-types to RAxML because we read in a plain RNA alignment and then need to tell RAxML by an additional text file that is passed via -S which RNA alignment sites need to be grouped/evolve together。
(37) – t 指定一个开始树的名字。树的格式要求是Newick格式。
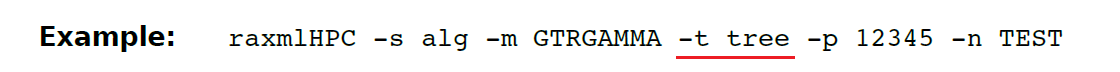
(38)- T (PTHREADS VERSION ONLY)仅仅限于多线程版本的RaxML。T的默认值是0.
用多少线程取决于你的CPUs。
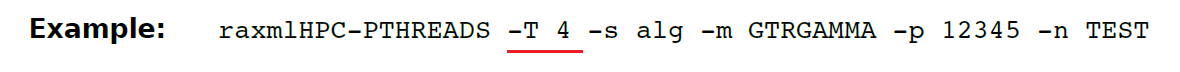
(39)- u 使用中位数表示GAMMA变异速率模型的近似离散。默认关闭。
(40)- U 尝试通过在大型gappy对齐上使用基于SEV的实现来节省内存。
(41)-v 显示版本信息。
(42)-V(大写)禁用变异速率模型。默认值:启用变异速率模型。
(43)-w(小写) 指定输出文件的路径。默认值是当前路径。
(44)-W(大写) 放置偏差算法的位点数,要和-f S(大写) 合用才行,默认是100 sites.
(45)-x 指定一个整数(随机数)并启用快速bootstrapping,与先前版本的RAxML不同,通过-m指定的变异速率性模型下进行快速BS replicates,而不是默认的在CAT下。
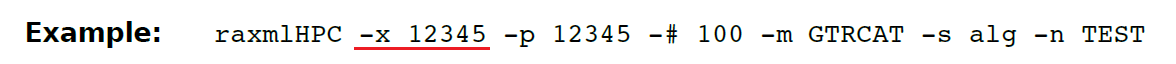
(46)-X(大写) 重建而不执行任何额外的SPRs。 这对于非常广泛的整个基因组数据集可能是有帮助的,因为这可以在拓扑上产生更多不同的起始树。
(47)- y 只用RAxML计算简约起始树,程序将在计算起始树后退出。默认值:关闭。
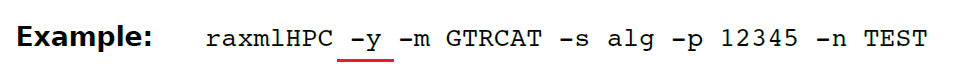
(48)- Y 极少用到。
(49)-z 指定包含多个树的文件的文件名,例如从一个bootstrap用于将二分值绘制到通过-t提供的树上。
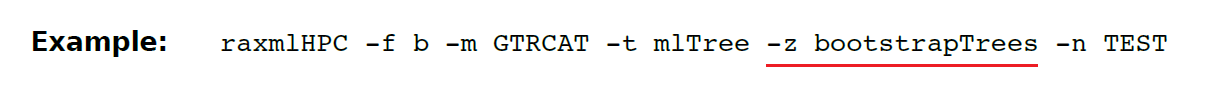
(50)-#或-N 指定不同起始树上的运行数,即设置Bootstrapping数值,现在论文一般要求1000次自举。将调用多个bootstrap分析。最好使用-N,-#可能会引起错误。默认值是1.
使用自举分析,先要将MPI版本的文件编译的(已经完成)。
(51) —mesquite 打印可由Mesquite解析的输出文件。默认值是关闭。

(52)—noseqcheck 禁用核对输入MSA的相同序列和完全未确定的位点。 启用此选项可以节省时间,特别是对于大型系统发育基因比对。在使用此选项之前,请确保使用-f c选项检查对齐!
(53)—**nobfgs 禁用自动使用BFGS方法以优化未分区DNA数据集上的GTR速率。 使用BFGS可以将模型优化的速度提高多达30%。 在分析单分区DNA数据集时,**默认情况下启用它。

(54)—epaprobthreshold=threshold 。默认值0.01.
specify a percent threshold for including potential placements of a read depending on the maximum placement weight for this read. If you set this value to 0.01 placements that have a placement weight of 1 per cent of the maximum placement will still be printed to file if the setting of epakeepplacements allows for it。
Here, RAxML will print those placements to the .jplace placement file that have a likelihood weight of at least 5% of that of the maximum placement weight for the specific read and if the number of placements to print as specified by epakeepplacements has not been exceeded
(55)—epaaccumulatedthreshold=threshold 指定累积似然权重阈值,其中将不同的读取位置打印到文件。 将打印读取的放置,直到其放置权重的总和达到阈值。 请注意,此选项既不能与epaprobthreshold结合使用,也不能与epakeepplacements结合使用!
上图的例子,在这里,RAxML会将读取的位置打印到.jplace放置文件,直到它们的累积似然权重达到0.95的值(总共1.0)。
四. 总结
到此为止,参数介绍已经完成。但是还有三个关键问题:
①应该使用哪个版本?
②实际中真正要设置的参数是那些?
③输出的结果哪个文件是树文件?
答:①②,参数设置不当会导致速度非常慢,基本我们会在服务器上跑数据,所以用到会是多线程,所以可以参考下面这个参数设置。
Linux 按 CPU 的计算速度,有 3 种版本: 标准版, SSE3 和 AVX 版本。 按并行化方式有 4 种版本: Sequential, Pthreads, MPI, Hybrid Pthreads/MPI 。可以编译出 12 个不同版本的 RAxML 程序。 SSE3 版本比标准版快 ~40%, AVX 版本比 SSE3 版本快 10~30% 。 与 CPU 支持有关
关于-HYBRID版本,前面已经提到,联合了MPI版本和PThreads版本,先用PThreads去计算单一树的似然值,再用MPI去处理自举检验或者对不同的共享的储存节点进行独立树的搜寻。
具体哪个版本快,没有明显的数据,我在使用中发现下面这个版本组合比较快:
./raxmlHPC-PTHREADS-AVX -s seqname -n zzj -m GTRGAMMAI -T 40 -N 1000 -p 20170808 -f a -x 20170808
(如果安装软件后没有添加到环境路径,先要进入你原先编译软件的那个文件夹,再敲命令,记得在最前面加上 ./)
注意: 如果运行这个版本报错,请检查是否编译安装了这个版本 。
参数介绍:
-s 输出用于建树的序列名字。
-n 定义输出的树文件的名字。
-m 这是模型设置,具体的模型要看你的是什么模型,上面仅仅是示例。
-T 我设置了40个线程,前提是你的服务器支持。
-N 设置自举次数,一般论文要求1000。
-p(小写) 一个用于简约推断的随机数,随机数可以自己设,12345也可以。
-f a 快速自举分析
-x(小写) 指定一个整数(随机数,可以随便取比如12345)并启用快速bootstrapping。
-k(小写) 指定bootstrapped树应该输出分支长度。默认值是关闭的。
(请注意,上面是我们平时的一般建树,如果是多基因联合建树,即分区数据建树,参数会有变动)
③生成的文件
如果使用总结里代码和参数最终会生成5个文件,其中RAxML_info是日志,另外4个是树文件,对我们真正有用的是树文件RAxML_bipartitionsBranchLabels,里面是一棵带有自展值加枝长信息的最佳ML树。另外,日志RAxML_info文件的最后部分里会有这4个树文件的区别介绍。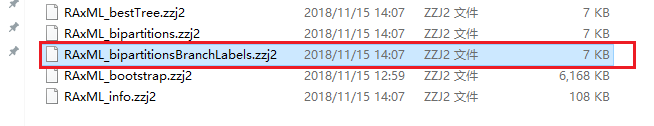
将打开方式选择“记事本”就可以查看这5个文件的内容。
如果你还添加了其他参数,可能会生成更多的文件。
④RaxML建树时会将高度相似的序列踢掉只留一条,其他不纳入建树分析,但是在最后生成的进化树上是有踢掉的序列名字的,这个不用担心。
转请注明出处。
如文中有错误之处,欢迎大家及时指正,一起交流学习。
By zzj
2018年11月16日初稿

若有收获,就点个赞吧
0 人点赞
