用过网页版本 BLAST 的童鞋都会发现,提交的序列比对往往在几分钟,甚至几十秒就可以得到比对的结果;而通过调用 API 却要花费几十分钟或者更长的时间!这到底是为什么呢?
NCBIWWW 基本用法
首先,我们来看一下提供了基于 API 在线比对的 Biopython 模块。
Biopython 中的 BLAST 提供了 over the Internet 和 locally 两种选择:Bio.Blast.NCBIWWW 主要是基于 NCBI BLAST API 用于在线比对;Bio.Blast.Applications 模块则是调用本地安装好的 BLAST 程序以及数据库执行比对。在这里我们来重点看一下 Bio.Blast.NCBIWWW 。
Bio.Blast.NCBIWWW 模块中主要是通过 qblast() 函数来调用 BLAST 的在线版本。它具有三个非可选参数:
- 第一个参数是用于搜索的 blast 程序,为小写字符串。目前,
qblast(biopython==1.7.4)仅适用于 blastn,blastp,blastx,tblast 和 tblastx。 - 第二个参数指定要搜索的数据库。关于这个选项,在 NCBI Guide to BLAST 上有详细的描述。
- 第三个参数是包含查询序列的字符串。这可以是序列本身,也可以是 fasta 格式的序列,或者是诸如 GI 号之类的标识符。
qblast 函数还接受许多其他选项参数,这些参数基本上类似于我们可以在 BLAST 网页上设置的不同参数。我们在这里只重点介绍其中一些:
- 参数
url_base是设置用于在 Internet 上运行 BLAST 的基本 URL。默认情况下,它连接到 NCBI(即 url_base=’https://blast.ncbi.nlm.nih.gov/Blast.cgi‘),但是可以使用它连接到云端运行的 NCBI BLAST 实例。更多详细信息请参阅 qblast 功能的文档。 - qblast 函数可以返回各种格式的 BLAST 结果,您可以使用可选的
format_type关键字进行选择:“HTML”,“Text”,“ASN.1” 或 “XML”。 默认值为 “XML”,因为这是解析器期望的格式。 - 参数
expect用于设置期望值或 e-value 阈值。
有关可选的 BLAST 参数的更多信息,请参考 NCBI 自己的文档或 Biopython 内置的文档:
>>> from Bio.Blast import NCBIWWW>>> help(NCBIWWW.qblast)Help on function qblast in module Bio.Blast.NCBIWWW:qblast(program, database, sequence, url_base='https://blast.ncbi.nlm.nih.gov/Blast.cgi', auto_format=None, composition_based_statistics=None, db_genetic_code=None, endpoints=None, entrez_query='(none)', expect=10.0, filter=None, gapcosts=None, genetic_code=None, hitlist_size=50, i_thresh=None, layout=None, lcase_mask=None, matrix_name=None, nucl_penalty=None, nucl_reward=None, other_advanced=None, perc_ident=None, phi_pattern=None, query_file=None, query_believe_defline=None, query_from=None, query_to=None, searchsp_eff=None, service=None, threshold=None, ungapped_alignment=None, word_size=None, alignments=500, alignment_view=None, descriptions=500, entrez_links_new_window=None, expect_low=None, expect_high=None, format_entrez_query=None, format_object=None, format_type='XML', ncbi_gi=None, results_file=None, show_overview=None, megablast=None, template_type=None, template_length=None)BLAST search using NCBI's QBLAST server or a cloud service provider.Supports all parameters of the qblast API for Put and Get.Please note that BLAST on the cloud supports the NCBI-BLAST CommonURL API (http://ncbi.github.io/blast-cloud/dev/api.html). Touse this feature, please set url_base to'http://host.my.cloud.service.provider.com/cgi-bin/blast.cgi' andformat_object='Alignment'. For more details, please seehttps://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastDocs&DOC_TYPE=CloudBlastSome useful parameters:- program blastn, blastp, blastx, tblastn, or tblastx (lower case)- database Which database to search against (e.g. "nr").- sequence The sequence to search.- ncbi_gi TRUE/FALSE whether to give 'gi' identifier.- descriptions Number of descriptions to show. Def 500.- alignments Number of alignments to show. Def 500.- expect An expect value cutoff. Def 10.0.- matrix_name Specify an alt. matrix (PAM30, PAM70, BLOSUM80, BLOSUM45).- filter "none" turns off filtering. Default no filtering- format_type "HTML", "Text", "ASN.1", or "XML". Def. "XML".- entrez_query Entrez query to limit Blast search- hitlist_size Number of hits to return. Default 50- megablast TRUE/FALSE whether to use MEga BLAST algorithm (blastn only)- service plain, psi, phi, rpsblast, megablast (lower case)This function does no checking of the validity of the parametersand passes the values to the server as is. More help is available at:https://ncbi.github.io/blast-cloud/dev/api.html
请注意,NCBI BLAST 网站上的默认设置与 qblast 上的默认设置不太相同。如果获得不同的结果,则需要检查参数(例如,e-value 值和 gap 值)。
例如,如果您要使用 BLASTN 在核苷酸数据库(nt)中搜索核苷酸序列,并且知道查询序列的 GI 号,则可以使用:
>>> from Bio.Blast import NCBIWWW>>> result_handle = NCBIWWW.qblast("blastn", "nt", "8332116")
另外,如果我们的查询序列已经存在于 FASTA 格式的文件中,则只需打开文件并以字符串形式读取此记录,然后将其用作查询参数:
>>> from Bio.Blast import NCBIWWW>>> fasta_string = open("m_cold.fasta").read()>>> result_handle = NCBIWWW.qblast("blastn", "nt", fasta_string)
我们还可以将 FASTA 文件作为 SeqRecord 对象进行读取,然后仅提供序列本身进行比对:
>>> from Bio.Blast import NCBIWWW>>> from Bio import SeqIO>>> record = SeqIO.read("m_cold.fasta", format="fasta")>>> result_handle = NCBIWWW.qblast("blastn", "nt", record.seq)
仅提供序列意味着 BLAST 将自动为您的序列分配一个标识符。您可能更喜欢使用 SeqRecord 对象的 format 方法来制作 FASTA 字符串(其中将包含现有标识符):
>>> from Bio.Blast import NCBIWWW>>> from Bio import SeqIO>>> record = SeqIO.read("m_cold.fasta", format="fasta")>>> result_handle = NCBIWWW.qblast("blastn", "nt", record.format("fasta"))
无论给 qblast() 函数提供什么参数,都应在 handle 对象(默认为 XML 格式)中返回结果。下一步是将 XML 输出解析为表示搜索结果的 Python 对象,但是您可能想先保存输出文件的本地副本。在调试从 BLAST 结果中提取信息的代码时,我发现这特别有用(因为重新运行在线搜索速度很慢,并且浪费了 NCBI 计算机时间)。
我们需要小心一点,因为我们只能使用 result_handle.read() 读取一次 BLAST 输出——再次调用 result_handle.read() 会返回一个空字符串。
>>> with open("my_blast.xml", "w") as out_handle:... out_handle.write(result_handle.read())...>>> result_handle.close()
完成上面的操作后,结果将保存在文件 my_blast.xml 中,并且原始句柄已提取了所有数据(因此我们将其关闭了)。 但是,BLAST 解析器的解析功能采用了类似于文件句柄的对象,因此我们可以打开保存的文件进行输入:
>>> result_handle = open("my_blast.xml")
现在我们已经将 BLAST 结果重新放回了句柄中,下一步,如果我们准备对它们进行处理,我们可以参考 Biopython 中 Parsing BLAST output 部分的内容,这里不再说明。
NCBIWWW 实现
在了解 NCBIWWW 的实现前,我们先来看一下 NCBI BLAST 对于 API 使用的一些说明:
https://blast.ncbi.nlm.nih.gov/Blast.cgi?CMD=Web&PAGE_TYPE=BlastDocs&DOC_TYPE=DeveloperInfo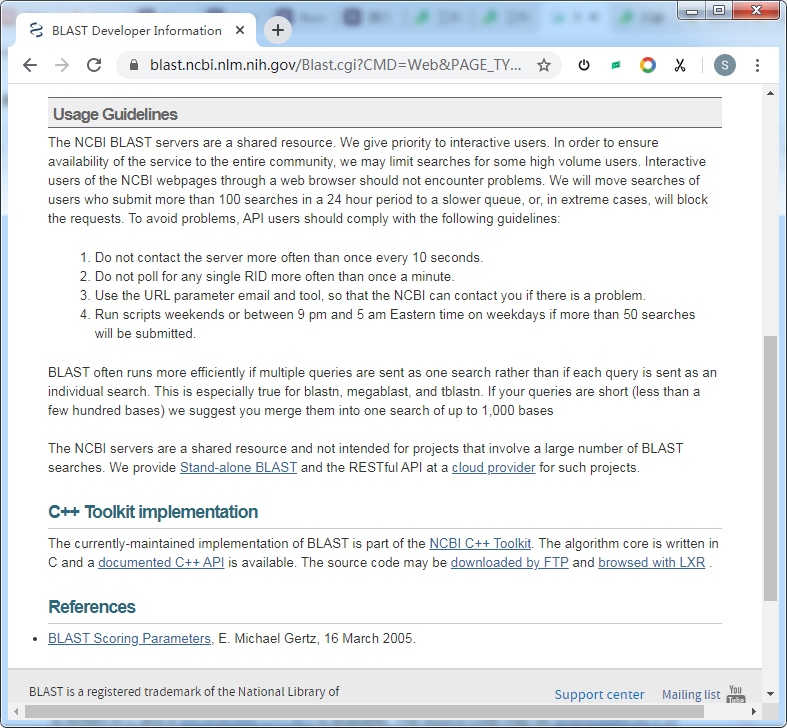
- NCBI BLAST 服务器是共享资源。为了确保整个社区都能使用该服务,他们可能会限制某些高流量用户的搜索。
- 他们会将在 24 小时内提交 100 次以上搜索的用户的搜索移到较慢的队列中,或者在极端情况下将阻止请求。
- NCBI BLAST 优先考虑互动的用户,通过网络浏览器的 NCBI 网页的交互式用户不会遇到以上的问题。
对于 API 的使用准则:
- 与服务器联系的频率不要超过每 10 秒一次。
- 不要轮询每一个 RID(Request ID) 多于一分钟一次。
- 使用 URL 参数电子邮件和工具,以便 NCBI 在出现问题时可以与您联系。
- 如果将提交超过 50 个搜索,则在周末或东部时间东部时间晚上 9 点至凌晨 5 点之间运行脚本。
我们再来看一下 NCBIWWW 在源码层面的处理: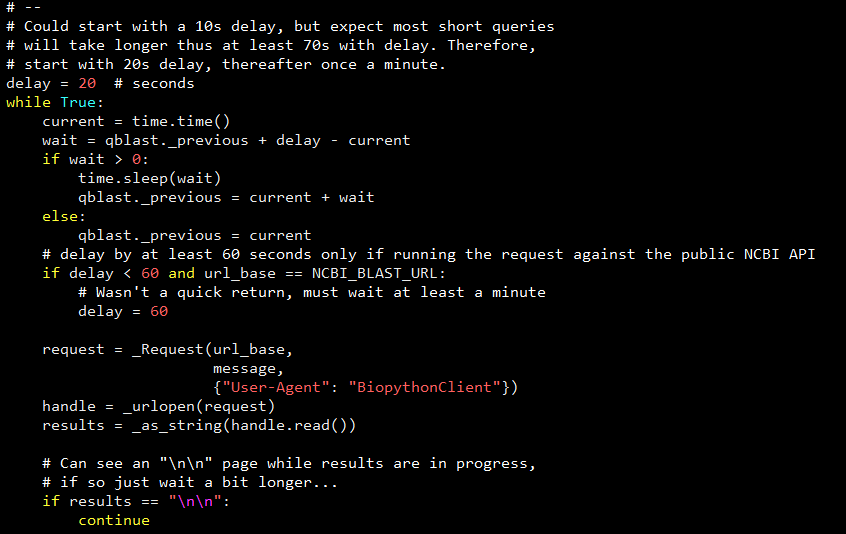
可以看到 NCBIWWW 从 20秒的延迟开始,然后开始每隔一分钟执行一次 request 轮询,直至任务完成或者任务出现异常。
所以,总的来说,NCBI BLAST API 的使用准则,加上 NCBI BLAST 对用户请求的任务队列处理,甚至 NCBI BLAST 服务器共享资源的限制,以及总用户请求数,这些都可能成为 NCBIWWW.qblast() 异常耗时的原因,这其中还不算个人服务器的网络影响。综上种种原因,如果考虑使用 NCBIWWW.qblast() 执行频繁的序列在线批处理,或许不是一个好的解决方案。
最后,基于 Python 的 NCBI BLAST 在线批处理,如果你有更好的方法,欢迎留言交流。

若有收获,就点个赞吧
0 人点赞
