1. channels 使用
需要注意的是做生信分析的童鞋使用 conda 环境时一定要特别注意 conda channels 的设置,滥用 channels 很有可能会导致你的软件升降级(甚至环境)错乱。推荐设置如下(~/.condarc):
channels:- https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/conda-forge- https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/bioconda- https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/main- https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/free- https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/r- https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/pro- defaultsshow_channels_urls: true
- mirrors.tuna.tsinghua.edu.cn/anaconda 是清华大学提供了一个 conda 的镜像;
- mirrors.ustc.edu.cn/anaconda 是中科大 conda 镜像,有需要的也可以使用;
- 生物信息软件 channel: mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/bioconda。
从 2019.04 起清华大学和中科大宣布停止 Anaconda 镜像服务,但是出于教育科研机构使用的前提,在 2019-05-15 清华大学又宣布重新恢复了 Anaconda 镜像!
因此原来使用国内镜像的 conda 可以根据自身需求决定是否需要变更新的 channels:
channels:- conda-forge- bioconda- main- free- r- pro- defaultsshow_channels_urls: true
更多 Anaconda channels,可以参考:https://repo.continuum.io/pkgs/。
2. 软件安装使用
conda 环境下的软件尽量使用 conda、pip 命令去安装。但同时也产生了一个问题,即 conda 中安装了 R,有些使用了 install.packages() , install_github , biocLite 等方式安装的 R 包,在环境迁移的时候,这些包如何迁移?
3. 环境激活
conda 4.6.x 切换环境使用的是:
$ source activate bio-base
conda 4.7.x 后切换环境变成了:
# To activate this environment, use:> conda activate bio-base# To deactivate an active environment, use:> conda deactivate
问题是,conda-4.7.x 使用推荐的命令切换环境,还要你 init 初始化一下 conda,不想 init 的话可以用回 4.6.x 的方式切换环境:
$ conda --versionconda 4.7.5$ conda activate bio-baseCommandNotFoundError: Your shell has not been properly configured to use 'conda activate'.To initialize your shell, run$ conda init <SHELL_NAME>Currently supported shells are:- bash- fish- tcsh- xonsh- zsh- powershellSee 'conda init --help' for more information and options.IMPORTANT: You may need to close and restart your shell after running 'conda init'.$ source activate bio-base(bio-base) shenweiyan@ecs-steven 10:49:59 /home/shenweiyan$ which python/Bioinfo/Pipeline/SoftWare/Anaconda3/envs/bio-base/bin/python$ source deactivate bio-baseDeprecationWarning: 'source deactivate' is deprecated. Use 'conda deactivate'.shenweiyan@ecs-steven 11:03:40 /home/shenweiyan$ source activate bio-base(bio-base) shenweiyan@ecs-steven 11:03:50 /home/shenweiyan$ conda deactivate(bio-base) shenweiyan@ecs-steven 11:03:57 /home/shenweiyan$
4. 环境导出与恢复
使用 conda 命令安装的包,都可以使用下面的命令导出依赖包/环境并批量恢复:
# To create a requirements.txt file# Gives you list of packages used for the environmentconda list# Save all the info about packages to your folderconda list -e > requirements.txt# To export environment fileactivate <environment-name>conda env export > <environment-name>.yml# For other person to use the environmentconda env create -f <environment-name>.yml# Install via `conda` directly.# This will fail to install all dependencies. If one fails, all dependencies will fail to install.conda install --yes --file requirements.txt# To go around issue above, one can iterate over all lines in the requirements.txt file.while read requirement; do conda install --yes $requirement; done < requirements.txt
5. R 与 R 包安装
R Essentials 软件包包含 IRKernel 和 80 多种最流行的数据科学 R 软件包,包括 dplyr,shiny,ggplot2,tidyr,caret 和 nnet 等。
5.1 R 软件
conda 安装 R 有很多种方法,如可以通过 r=3.6.x,或者 r-base、r-irkernel、r-essentials 都可以。需要注意:
如果需要在 Anaconda 的 Jupyter Notebook 中使用 R,建议使用 conda install -c r r-irkernel 或者 conda install -c r r-essentials 的方式安装,因为 conda install -c r r=3.6.x/r-base 默认不会安装 irkernel,而且先安装的 r=3.6.x/r-base 可能与后安装的 r-irkernel/r-essentials 产生冲突。
5.2 R 包
通过 conda 安装的 R,在安装 R 包时,最好使用 conda 命令去安装,conda 无法安装的(如 github 的包)再考虑其他的安装方式。
1. install.packages
install.packages() 所有 R 包:
> data = read.table("r-packages.txt", header=T, check.names=F, quote="")> soft = as.vector(data[,1])> install.packages(soft)
2. bioconductor
Bioconductor 镜像:
Bioconductor 镜像源配置文件之一是
.Rprofile(linux 下位于~/.Rprofile)。在文末添加如下语句:#清华大学开源镜像options(BioC_mirror="https://mirrors.tuna.tsinghua.edu.cn/bioconductor")
也可以在安装过程中指定镜像:
source("http://bioconductor.org/biocLite.R")#指定一个离你最近的国内镜像options(BioC_mirror="http://mirrors.ustc.edu.cn/bioc/")biocLite("包名")
biocLite 包安装:
在 R-3.5(Bioconductor-3.7) 前,Bioconductor 都是通过 biocLite 安装相关的 R 包:
> source("http://bioconductor.org/biocLite.R")> options(BioC_mirror="http://mirrors.tuna.tsinghua.edu.cn/bioconductor")> data = read.table("r-biocLite.txt", header=T, check.names=F, quote="")> head(data)biocLite_Packages_Name1 RSQLite2 KEGGREST3 png4 Rcpp5 digest6 AnnotationDbi> soft = as.vector(data[,1])> biocLite(soft)
从 R-3.5(Bioconductor-3.8) 起,Bioconductor 开始使用 BiocManager 安装 R 包:
if (!requireNamespace("BiocManager", quietly = TRUE))install.packages("BiocManager")BiocManager::install()
3. github_install
GitHub 上的一些最新 R 包,可以使用 devtools 进行安装:
install.packages("devtools")library(devtools)install_github("jokergoo/ComplexHeatmap")
6. 特别注意的软件
6.1 gcc
对于使用 conda install --yes --file requirements.txt 重装某一个环境的所有软件时,如果软件中包含了 gcc,安装了 R 后,在使用 R 时会可能会引发错误:
/path/to/SoftWare/Anaconda/v2/lib/R/bin/exec/R: /path/to/SoftWare/Anaconda/v2/lib/R/bin/exec/../../lib/../../libgomp.so.1: version `GOMP_4.0' not found (required by /path/to/SoftWare/Anaconda/v2/lib/R/bin/exec/../../lib/libR.so)
6.2 glibc
glibc 是 GNU 发布的 libc 库,即 c 运行库。glibc 是 linux 系统中最底层的 api,几乎其它任何运行库都会依赖于 glibc。glibc 除了封装 linux 操作系统所提供的系统服务外,它本身也提供了许多其它一些必要功能服务的实现。由于 glibc 囊括了几乎所有的 UNIX 通行的标准,可以想见其内容包罗万象。而就像其他的 UNIX 系统一样,其内含的档案群分散于系统的树状目录结构中,像一个支架一般撑起整个作业系统。在 GNU/Linux 系统中,其 C 函式库发展史点出了 GNU/Linux 演进的几个重要里程碑,用 glibc 作为系统的 C 函式库,是 GNU/Linux 演进的一个重要里程碑。
有一些软件对于 glibc 的版本会有要求,如 cnvnator-0.3.3 要求 glibc >= 2.14。虽然在 anaconda 中有很多 channel 都提供了 glibc,但千万注意一定要注意不要轻易去安装,否则有很大的概率会导致整个环境挂掉,甚至会导致当前环境中的 conda、python 出现动态库混乱错误,恢复起来相当麻烦!
我在《一次”胆战心惊”的真实集群运维经历》记录了 gblic 的一些集群吐血经历,感兴趣的可以了解一下。
7. 什么时候使用 Anaconda
对于 Anaconda(conda) 软件安装以及依赖解决的原理,我对这个黑盒子表示一头雾水。真实的情况是,如果在一个环境中安装了几百个软件(包),再去新装软件,这时候 Anaconda 常常会卡在 Collecting package metadata 和 Solving environment 过程中,甚至一个晚上都没法解决环境的依赖。
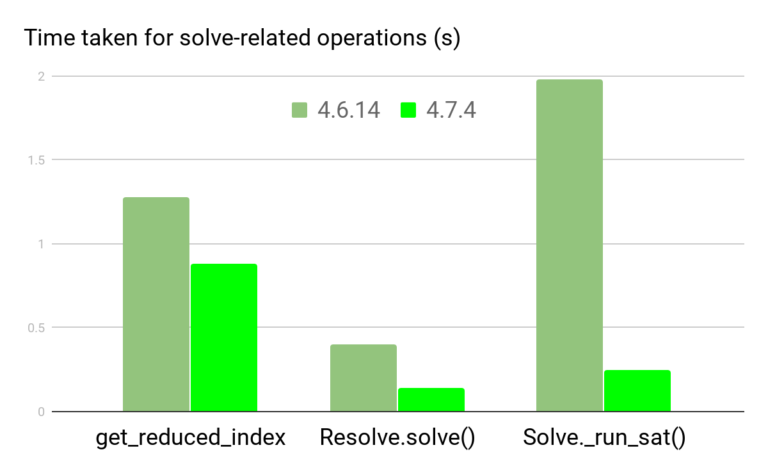
conda 官方说他们在 conda-4.7 中花了很多的精力去提升了 conda 的速度(参考官方文章:《How We Made Conda Faster in 4.7》),但从 4.6 升级到 4.7 过程很容易导致环境崩溃,修复起来极其麻烦(反正我折腾了一个晚上都没能把我的 base 给恢复回来,吐血的经历)!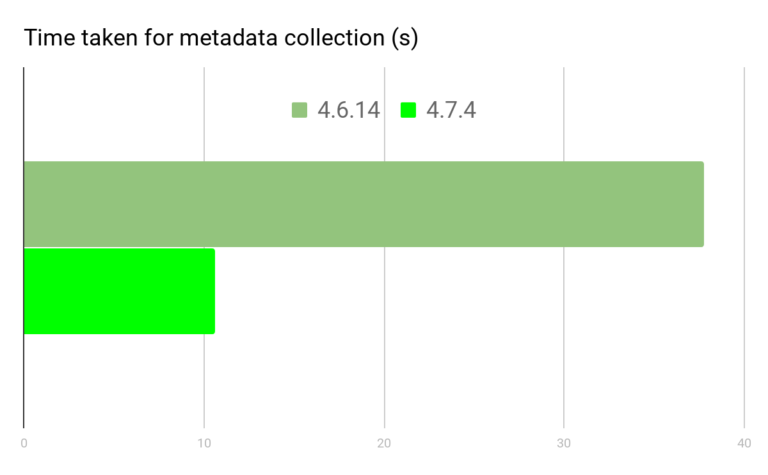
对于新手而言,Anaconda 的确是非常简单易用,如果对于多用户的服务器端,或者是提供公共使用的软件库是否需要采用 Anaconda,个人觉得的确需要慎重考虑一下,最起码需要考虑:
- 是否需要根据流程划分环境?如果每个流程都需要使用 Python+R+Perl,每个环境都安装这三个软件(包),如何避免空间浪费?
- 需要考虑如何备份和恢复环境。万一某个环境崩溃,有什么快速替代的方案而不影响业务。
或许还有更多的问题,这里没有列出来,如果大家有什么更好的想法或者建议,也欢迎留言交流。

若有收获,就点个赞吧
0 人点赞
