本文节选自《罕见病诊疗指南 2019年版》,全文下载: http://www.gov.cn/fuwu/2019-02/28/content_5369203.htm

《Gitelman综合征诊疗中国专家共识(2021版)》重磅出炉!请参考2021版
【GS】Gitelman 综合征诊疗中国专家共识(2021 版)
1 概述
Gitelman 综合征(Gitelman syndrome,GS;OMIM 263800)是一种由肾脏远曲小管钠氯协同转运蛋白(NCC)功能障碍所致的常染色体隐性遗传病。1966年由美国医生 Gitelman 首先报道了该病,但直至 1996 年其致病基因 SLC12A3才得以明确。主要临床特点为肾性失钾导致的低钾血症、代谢性碱中毒,常伴有低血镁、低尿钙和肾素-血管紧张素-醛固酮系统(RAAS)活化,血压正常或偏低。
2 病因和 流行病学
Gitelman 综合征是由编码噻嗪类利尿剂敏感的钠氯协同转运体(NCC)的SLC12A3 基因突变所致。生理情况下,通道蛋白 NCC 位于肾脏远曲小管上皮细胞的管腔侧,参与肾小球滤过液中 5%~10%氯离子和钠离子的重吸收,是机体维持水、电解质平衡的一道重要防线。当基因突变导致 NCC 结构和(或)功能障碍时,氯离子和钠离子从远端肾小管重吸收减少,肾脏重吸收水减少,继发性RAAS 活化、肾性失钾和钙重吸收减少。目前在 Gitelman 综合征患者中已发现近 500 种 SLC12A3 基 因 突 变 ( http://www.hgmd.cf.ac.uk/ac/gene.php?gene=SLC12A3)。此外,编码氯离子通道 ClC-Kb 的 CLCNKB 基因突变(Batter 综合征Ⅲ型)和编码肝转录因子 1-β(HNF1-β)的 HNF1B 基因突变也可产生类似临床表现。
Gitelman 综合征是最常见的遗传性肾小管疾病之一,患病率约为 1/40 000~1/4000,亚洲人群中可能更高。由于该病易被漏诊或误诊,很难确定一般人群中该病的真实患病率,目前没有观察到男性和女性发病率的显著差异。
3 临床表现
Gitelman 综合征常于青少年或成年早期起病。临床表现主要与低血钾和低血镁相关,轻型患者可无症状或表现为轻度乏力和纳差;严重患者会出现四肢抽搐、软瘫、痛性痉挛、晕厥和横纹肌溶解继发急性肾损伤,甚至因为严重室性心律失常导致心脏骤停。目前认为,Gitelman 综合征临床表现的异质性不仅与基因突变类型和修饰基因相关,还与患者性别和饮食习惯等环境因素相关。
常见临床表现如下:
- 全身症状 疲乏、口渴、多饮、嗜盐。
- 神经-肌肉系统 肌无力、痛性痉挛、抽搐、惊厥发作、肢体麻木、感觉异常、横纹肌溶解、瘫痪、头晕、眩晕、共济失调和假性脑瘤等。
- 心血管系统 心悸、晕厥、血压正常或偏低和室性心律失常等。
- 消化系统 便秘、呕吐等。
- 泌尿系统 多尿、夜尿增多、遗尿、蛋白尿和肾功能不全。
- 骨关节系统 关节痛、痛性痉挛(软骨钙质沉着症)。
- 内分泌和生长发育 生长迟缓、青春期延迟;长期低钾和低镁的患者糖尿病或者糖耐量减低的比例并不少见。
- 眼部症状 少数患者会出现视物模糊和巩膜脉络膜钙化。
Gitelman 综合征患者蛋白尿和肾功能损害的原因主要与长期低血钾相关,后者可导致肾小管间质损伤和囊肿形成,蛋白尿以小管来源为主;其次因 RAAS长期激活,可能直接或间接导致肾小球节段硬化,极少数病例合并有其他肾小球疾病,如 IgA 肾病,C1q 肾病等,但其与 Gitelman 综合征之间的相关性并不明确。
辅助检查
- 血清(浆)学检测 钾(K)、镁(Mg)、钙(Ca)、磷(P)、尿酸(UA)、肌酐(Cr)、血气、立位肾素-血管紧张素-醛固酮水平、血糖或糖耐量检查。
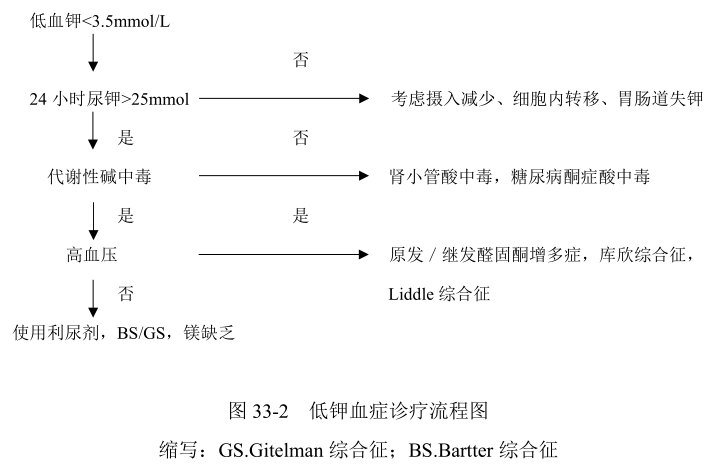
- 尿液检测 尿常规,24 小时尿蛋白定量,24 小时尿钾、钠、氯、镁、钙、磷、尿酸和肌酐;其中血钾小于 3.5mmol/L 时,24 小时尿钾大于 25mmol 可符合肾性失钾。
- 心电图 评估 QT 间期是否延长,是否合并心律失常等表现。
- 肾脏超声 肾脏形态多正常,长期低钾患者可出现肾囊肿,可用于排除其他因为肾脏结构异常导致的肾性失钾。
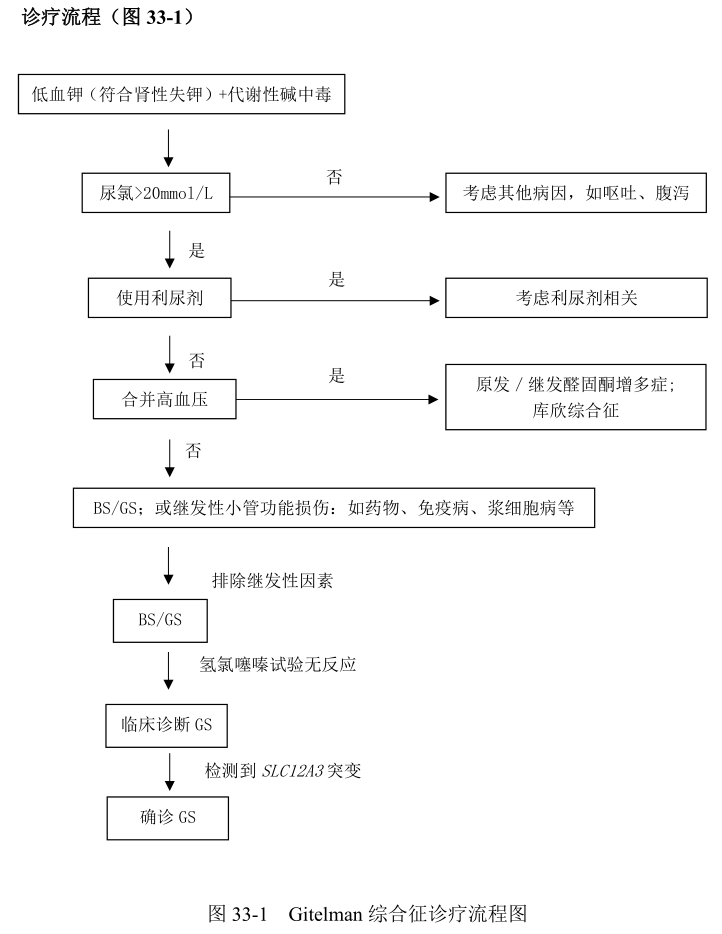
- 氢氯噻嗪试验:通过小剂量氢氯噻嗪(50mg)直接阻断 NCC,观察使用前后氯离子排泄分数的变化程度(△FECl),与正常对照进行比较,评估 NCC功能。中国人群中△FECl<2.86%时诊断 Gitelman 综合征的灵敏度和特异度分别为 95.7%和 95.8%。目前临床采用改良的氢氯噻嗪试验较为安全且简便易行,成本低,但对于怀疑 Batter 综合征的患者,需注意监测血钾进一步降低的风险。
- 基因检测 是诊断 Gitelman 综合征的金标准。检测到 SLC12A3 纯合突变或复合杂合突变可确诊,单杂合突变的患者需结合临床,新发现的突变需要体外功能试验确定突变的致病性。
诊断
- 慢性肾性失钾导致的低钾血症,伴有代谢性碱中毒,血浆肾素-血管紧张素-醛固酮系统活化(立位),但血压正常或偏低的患者,应考虑失盐性肾病。
- 如同时合并低血镁(<0.7mmol/L)或低尿钙(成人尿钙肌酐比<0.2mmol/mmol)应高度怀疑 NCC 功能障碍。进一步的氯离子清除试验(氢氯噻嗪试验)有助于定位 NCC 功能障碍,并能初步判断损害程度。需要注意的是,即使血镁正常或者尿钙不低,并不能除外 Gitelman 综合征。
- 需要除外长期服用氢氯噻嗪类利尿剂或缓泻剂,其他因为药物、免疫病(如干燥综合征)或者单克隆免疫球蛋白病等导致的远端小管功能障碍。
- 确诊需要基因诊断。
鉴别诊断
Gitelman 综合征主要与以下两类疾病鉴别:
其他原因的低钾血症 首先需要根据病史和24小时尿钾与血钾水平比较,确定是否肾性失钾。然后检测尿氯水平,如尿氯排泄水平不高(<20mmol/L=则需警惕呕吐、腹泻等情况,反之则考虑存在肾性失氯;合并高血压应警惕原发性/继发性醛固酮增多症,库欣综合征;如合并代谢性酸中毒要警惕肾小管酸中毒。其次除外其他药物(特别是利尿剂和中药)、免疫病和浆细胞病所继发的肾性失钾,可以通过相应的检查帮助鉴别。
其他失盐性肾病 如 Batter 综合征。经典 Batter 综合征(Batter 综合征Ⅲ型)由编码氯离子通道 ClC-Kb 的 CLCNKB 基因突变所致,其起病相对较早(3岁以前),低钾程度更重,更易出现生长迟缓、多尿,患者血镁水平多正常,尿钙水平正常或偏高。患者对氢氯噻嗪试验有反应,说明 NCC 功能正常;但呋塞米试验没有反应,有助于临床鉴别。基因检测有助于确诊。
治疗
Gitelman 综合征以对症治疗、电解质替代治疗为主,以期达到缓解症状、提高生活质量、避免严重并发症的目标。总体治疗原则如下:
- 替代治疗 推荐高盐饮食,进食富含钾、镁的食物,口服氯化钾、门冬氨酸钾镁、硫酸镁和氯化镁等药物,紧急或严重情况下可静脉输注钾盐和镁盐。2017年改善全球肾脏病预后组织(KDIGO)专家争议共识建议血钾和血镁治疗目标分别为 3.0mmol/L 和 0.6mmol/L。
- 其他治疗 保钾利尿剂(如螺内酯、依普利酮)、肾素-血管紧张素系统抑制剂(低血压时慎用)抑制 RAAS 活化,前列腺素合成酶抑制剂(吲哚美辛等)有助于减少补钾药物的剂量,改善低钾相关症状。但需注意监测相关药物副作用。
- 患者管理和宣教 强调个体化的疾病管理,培养和加强患者自我监测症状体征,按时使用药物、适时就医、规律随诊,并需要重视患者的心理健康。
- 特殊情况 对于妊娠期、围手术期及合并其他疾病的 Gitelman 综合征患者,应加强监测并积极随访,及时调整药物,避免病情加重及严重并发症。
参考文献
[1] Blanchard A, Bockenhauer D, Bolignano D, et al. Gitelman syndrome: consensus and guidance from a Kidney Disease: Improving Global Outcomes (KDIGO)Controversies Conference. Kidney Int, 2017,91:24-33.
[2] Gitelman 综合征诊治专家共识协作组. Gitelman 综合征诊治专家共识.中华内科杂志, 2017, 56(9):712-716.
[3] 彭晓艳,陈丽萌. Gitelman 综合征:从病理生理到临床实践. 国际药学研究杂志, 2017,44(2):157-166.
[4] 彭晓艳, 蒋兰萍, 袁涛,等. 氯离子清除试验在 Gitelman 综合征鉴别诊断中的应用. 中国医学科学院学报, 2016,38(3):8.
氯离子清除试验在Gitelman综合征鉴别诊断中的应用.pdf
[5] Ma J,Ren H,Lin L, et al. Genetic features of Chinese patients with Gitelman syndrome: sixteen novel SLC12A3 mutations identified in a New Cohort.Am J Nephrol,2016,44(2):113-121.
Genetic features of Chinese patients with Gitelman syndrome-sixteen novel SLC12A3 mutations identified in a New Cohort.pdf
[6] Liu T,Wang C,Lu J, et al. Genotype/Phenotype analysis in 67 Chinese patients with Gitelman’s syndrome. Am J Nephrol,2016,44(2):159-168.
Genotype Phenotype analysis in 67 Chinese patients with Gitelman’s syndrome.pdf
[7] Jiang L, Chen C, Yuan T, et al. Clinical severity of Gitelman syndrome determined by serum magnesium. Am J Nephrol,2014,39:357-366.
Clinical severity of Gitelman syndrome determined by serum magnesium.pdf

若有收获,就点个赞吧
0 人点赞
