作者: 中国研究型医院学会罕见病分会,中国罕见病联盟,北京罕见病诊疗与保障学会 Gitelman 综合征诊疗中国专家共识(2021 版)[J/OL].协和医学杂志.
中国研究型医院学会罕见病分会,中国罕见病联盟,北京罕见病诊疗与保障学会,
陈丽萌1 ,魏 珉 2 ,刘晓清 3,4 ,刘雅萍 5 ,夏维波 6 ,刘俊涛 7 ,李雪梅 1 ,张抒扬 8
中国医学科学院,北京协和医学院,北京协和医院 1 肾内科 2 儿科 3 感染内科 4 临床流行病学教研室 6 内分泌科 7 妇产科 8 疑难重症及罕见病国家重点实验室,北京 100730
5 中国医学科学院基础医学研究所医学遗传学系,北京 100730
通信作者:张抒扬 电话:010-69155810,E-mail:shuyangzhang103@163.com
DOI:10.12290/xhyxzz.2021-0555
Gitelman综合征诊疗中国专家共识(2021版).pdf
摘要
近年来,国内对 Gitelman 综合征的研究取得了长足进步,中国人群 Gitelman 综合征新的临床表型、疾病分型、改良功能试验、生物标志物和特殊人群管理经验相继在国内外期刊报道。基于最新循证医学证据,多学科领域专家在 Gitelman 综合征的临床表型、诊断、治疗、慢性并发症及合并症管理方面达成一致共识,为 Gitelman 综合征的规范化诊断和治疗提供了重要依据。
关键词
Gitelman综合征;诊断;治疗
Gitelman 综合征(Gitelman syndrome,GS)是一种罕见的遗传性肾小管疾病,其临床特点主要为低钾血症、代谢性碱中毒、低镁血症、低钙尿症[1-2] 。GS 的遗传方式为常染色体隐性遗传,由编码位于肾远曲小管的噻嗪类利尿剂敏感的钠氯共转运蛋白(sodium-chloridecotransporter,NCC)基因 SLCl2A3 发生变异所致。GS 的患病率约为 1~10/4 万,亚洲人群可能更高[1-2] 。2017 年,改善全球肾脏病预后组织(Kidney Disease:Improving Global Outcomes,KDIGO)制定了首个“GS 争议共识和指导意见”[1] ;同年 9 月,中国 GS 诊治专家共识协作组发布了中国《Gitelman 综合征诊治专家共识》[2] 。2018 年,国家卫生健康委员会、科技部、工信部、国家药品监督管理局、国家中医药管理局等五部委联合制定的《第一批罕见病目录》将 GS 纳入其中。2019 年,受国家卫生健康委员会委托,国家卫生健康委罕见病诊疗与保障专家委员会办公室牵头制定了我国首部《罕见病诊疗指南(2019)》,涵盖GS 在内的 121 种罕见病。这些共识和指南对于规范 GS 的诊治发挥了巨大推动作用,国内 GS临床研究也取得了长足进步,关于中国人群 GS 新的临床表型、分型、改良功能试验、生物标志物和特殊人群管理的经验相继在国内外期刊被报道。为进一步明确 GS 的临床表现,规范诊断、治疗和管理策略,中国研究型医院学会罕见病分会、中国罕见病联盟、北京罕见病诊疗与保障学会联合相关领域专家,以循证医学为基础,制订本共识,旨在帮助临床医师制订规范化的 GS 诊疗策略。
1 共识制订方法
2020 年 7 月 20 日,“Gitelman 综合征诊疗专家共识研讨会”召开,多学科专家讨论了共识的总体框架,并进行具体任务分工;基于标准的疾病模式(临床表现、诊断、疾病分级与分型、治疗),并结合 GS 研究最新进展,确定了共识拟解决的问题;组建了共识制订工作组,包括专家组、秘书组和编写组,其中专家组成员来自肾内科、儿科、内分泌科、产科、麻醉科、检验科、遗传学和临床流行病学领域。
共识编写组以“Gitelman syndrome”“Gitelman 综合征”“吉特曼综合征”为主题词检索了 PubMed、Embase、Cochrane Library、ClinicalTrials、中国知网、万方数据知识服务平台和中文维普数据库,检索时间为建库至 2020 年 7 月 10 日,经去重处理后共获得相关文献 1634 篇,阅读文献摘要和/或全文,最终纳入符合本共识主题的文献 76 篇。依据牛津循证医学中心证据分级方法并参考国内外共识制订的方法学文献[3-4] ,将纳入的研究证据分为 6 个证据等级(1a、1b、2a、2b、3、4)(表 1),并根据证据等级将推荐强度分为 A(强)、B(中)、C(弱)3 级(表 2),以代表共识制订专家组的建议。2021 年 5 月初,基于国内外最新研究成果,编写组完成了共识意见初稿;5 月 15 日,于北京举行了共识专家组会议;7 月5 日,专家组进行线上交流、沟通,逐条讨论、修改和完善共识意见;7 月 23 日,所有条款均经专家组无记名投票,赞成人数在 85%以上被认为专家意见达成共识[5] ,最终形成共识终稿。
【表1】本共识的证据等级定义
| 证据等级 | 定义 |
|---|---|
| 1a | 总结多项随机对照研究的荟萃分析 |
| 1b | 至少有一项随机对照研究 |
| 2a | 至少有一项设计良好的对照研究,但未随机分组 |
| 2b | 至少有一项设计良好的其他类型的准试验研究 |
| 3 | 设计良好的非试验性描述性研究,如比较研究、相关性研究、病例研究等 |
| 4 | 专家委员会的报告或观点,以及权威专家的临床经验 |
【表2】本共识的推荐强度定义
| 推荐强度 | 定义 | 证据等级 |
|---|---|---|
| A(强) | 有一项或多项高质量的随机对照研究回答该临床问题 | 1a、1b |
| B(中) | 针对该问题,有设计良好的临床研究但未随机分组 | 2a、2b、3 |
| C(弱) | 专家委员会的报告或观点,以及权威专家的临床经验, 提示本领域需要进行高质量的临床研究 |
4 |
2 临床表现与诊断
2.1 临床表现
GS 常见临床症状为低血钾、低血镁在全身多系统的表现,常累及骨骼肌、肾脏、胃肠道、心血管和神经系统,来自中国人群的报告数据见表 3[6] 。儿童 GS 患者常见就诊原因为手麻、肌无力、生长发育迟缓、手足抽搐等。大样本量病例报告显示 70% 的 GS 患者同时存在至少3个系统的临床症状[7] 。GS 患者还可出现蛋白尿和肾功能损害,肾脏病理除球旁器增生外,还可表现为慢性肾小管间质损伤[1] ,少数病例合并有其他肾小球疾病。GS 合并肾小球损害的病例包括局灶节段性肾小球硬化[8] 、C1q 肾病 [9-10] 、微小病变肾病 [11] 等,但目前尚不能在二者之间建立因果关系。
【表3】中国人群 Gitelman 综合征患者的临床表现
| 类别 | 常见症状(>50%) | 多见症状(20%~50%) | 偶见症状(<20%) |
|---|---|---|---|
| 全身症状 | 嗜盐,疲乏 | 口渴,多饮 | - |
| 神经-肌肉系统 | 肌肉无力 | 手足抽搐、肌肉僵硬疼 痛,感觉异常,头晕 |
眩晕,共济失调,软 瘫,呼吸困难 |
| 心血管系统 | 心悸,血压偏低 | QT 间期延长 | 晕厥 |
| 泌尿系统 | 夜尿增多 | 多尿 | - |
| 骨关节系统 | - | 软骨钙化 | 关节痛 |
| 消化系统 | - | - | 腹痛 |
2.2 临床诊断、 基因诊断与功能诊断
推荐意见:
- (1)对于青少年或成年发病,表现为代谢性碱中毒、血压正常的低钾血症、低镁血症或低钙尿症患者,应排查 GS(证据等级:3;推荐强度:B)。
- (2)氢氯噻嗪试验是辅助诊断 GS 的一种重要临床功能试验,有助于鉴别肾小管损伤部位,反映肾脏钠氯共转运蛋白功能状态(证据等级:3;推荐强度:B)。
- (3)建议临床诊断为 GS 的患者行基因检测确认,GS 高频突变可能存在种族差异(证据等级:3;推荐强度:B)。
- (4)最常用的基因检测方法是 SLC12A3 基因直接测序法,而二代测序、多重连接探针扩增技术(multiplex ligation-dependent probe amplification,MLPA)也逐渐用于 GS 诊断(证据等级:3;推荐强度:B)。
- (5)根据 ACMG 基因变异致病性解读指南对变异进行分类,必要时可开展体内或体外功能试验,为确定变异致病性提供证据(证据等级:3;推荐强度:B)
2.2.1 临床诊断
- 根据病史除外消化道钾摄入不足或腹泻、使用利尿剂、细胞内外钾分布异常等情况。
- 存在肾性失钾及低钾血症相关临床表现,可伴有低镁血症或低钙尿症。肾性失钾:血钾<3.0 mmol/L 时,尿钾排泄量>20 mmol/24 h;或血钾<3.5 mmol/L 时,尿钾排泄量>25 mmol/24 h。
- 血压正常或偏低。
- 代谢性碱中毒。
2.2.2 Bartter综合征的鉴别诊断
经典的 Bartter 综合征(Bartter 综合征Ⅲ型)由编码氯离子通道 ClC-Kb 的 CLCNKB 基因突变所致[12] ,患者发病相对较早(多在 3 岁前),更易出现生长发育迟缓,血镁水平多正常,尿钙水平正常或偏高。行氯离子清除试验,若患者对氢氯噻嗪试验有反应而对呋塞米试验无反应,将有助于临床诊断 Bartter 综合征,基因检测可进一步鉴别。Gitelman 综合征具体诊疗流程见图 1。
【图1】Gitelman 综合征诊疗流程图
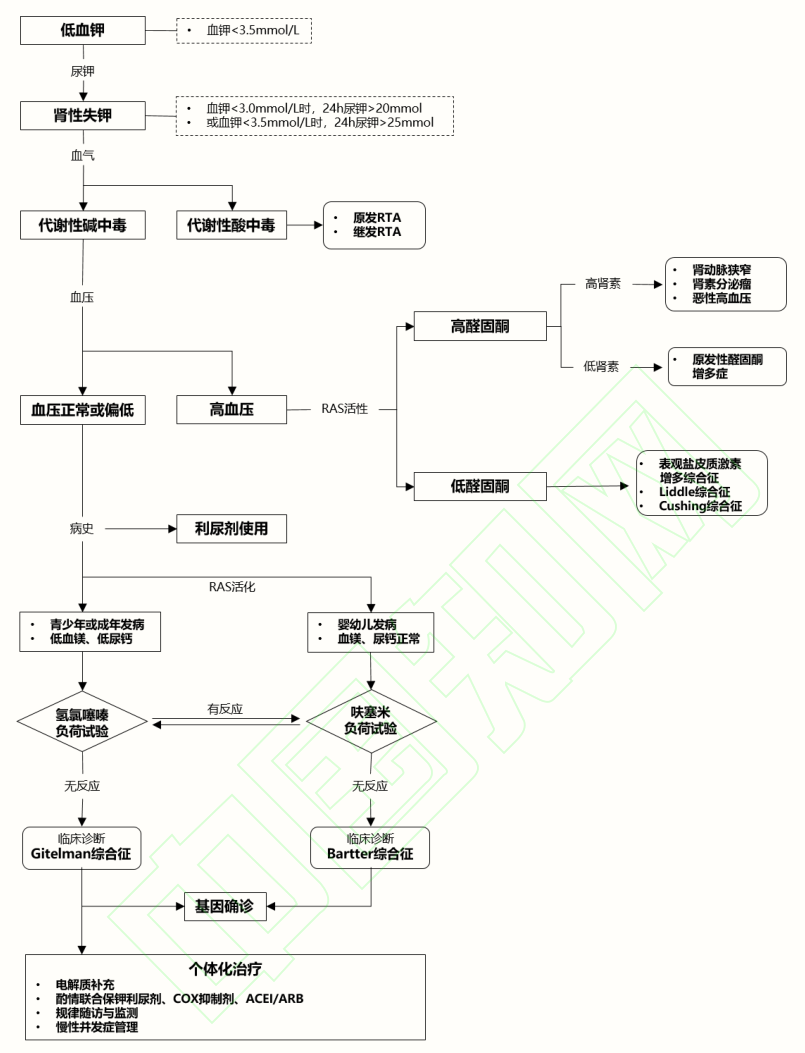
RTA:肾小管酸中毒;RAS:肾素-血管紧张素系统;COX:环氧化酶抑制剂;ACEI/ARB:血管紧张素转 化酶抑制剂/血管紧张素受体拮抗剂
2.2.3 基因诊断
2.2.3.1 高频突变
目前已知的 SLC12A3 基因突变超过 500 个(人类基因突变数据库:http://www.hgmd.org)。中国人群突变频率较高的 2 种基因突变为 p.T60M 和 p.D486N[7,13-19] ,欧洲人群突变频率较高的 7 种基因突变为 p.A313V、c.1180+1G>T、p.G741R、p.L859P、p.R861C、c.2883+1G>T 和 p.C994Y[20] 。两组人群的高频突变无重叠,提示 GS 的基因突变分布可能存在种族差异。
2.2.3.2 基因检测策略
针对 SLC12A3 基因的直接测序仍是目前使用最广泛的检测方法,但约 8%~30%的患者仅可检测到单杂合突变[7,19-22] ,需进一步对内含子突变 [23-25] 及基因大片段缺失和重复 [18] 进行分析。二代测序技术、MLPA 和微阵列比较基因组杂交技术(array-based comparative genomichybridization,aCGH)逐渐用于诊断 GS[20,24,26-29] 。对一代测序仅有SLC12A3 单杂合突变的患者,建议可进一步行 MLPA、全外显子组或全基因组二代测序寻找其他可能的变异位点,如条件允许可直接采用二代测序技术进行基因诊断。
2.2.3.3 基因诊断解读
SLC12A3 纯合突变或复合杂合突变可确诊 GS,单杂合突变的患者需结合临床情况进行分析。发现基因变异后,可在 1000 Genomes、GnomAD 等数据库中查询该变异在正常人群中出现的频率,在人类基因突变数据库查阅或检索文献明确是否为已报道变异,并根据 ACMG 基因变异致病性解读指南对其进行分类[30] 。必要时可开展体内外功能试验,为确定变异致病性提供证据。爪蟾卵母细胞表达体系是目前国际公认的验证错义变异对 NCC 功能影响的最好体外模式细胞方法[21,31-34] ,氢氯噻嗪试验也有助于评估患者体内的 NCC 功能水平。
2.2.4 功能诊断
氯离子清除试验在 GS 鉴别诊断中具有重要意义。应用小剂量可直接抑制 NCC 和 Na-K-2Cl 共转运蛋白功能的药物——氢氯噻嗪和呋塞米,进行氯离子清除试验(临床生理功能试验),有助于鉴别 GS 和 Bartter 综合征[35] 。
2.2.4.1 原理与方法
通过小剂量氢氯噻嗪(成人 50mg,7~17 岁 1mg/kg)直接阻断 NCC,观察服药前后氯离子排泄分数(fractional excretion,FE)的变化程度以评估 NCC 功能。由于 NCC 功能缺陷,GS 患者氢氯噻嗪试验结果通常表现为反应性降低,其降低程度可间接反映 NCC 功能损伤的严重程度。国内应用较多的氢氯噻嗪试验流程共需 4h(图 2),试验前患者需停止补钾、补镁治疗 1d,且保证试验前血钾水平不低于 3 mmol/L,试验过程中需定时饮水、留取尿标本、采集血液标本。
氯离子排泄分数计算方法为:FEcl = 100×(UCl / SCl )×(SCR / UCR )
( FEcl 为氯离子排泄分数,UCl 为尿氯离子浓度, SCl 为血氯离子浓度,SCR为血肌酐浓度,UCR为尿肌酐浓度)。
【 图2】改良的氯离子清除试验流程图

2.2.4.2 诊断价值及安全性
氢氯噻嗪试验对于 GS 诊断的临床价值已在不同人种中得以证实[22,35-37] 。陈丽萌等 [36] 首次将改良的氯离子清除试验方法应用于中国 GS 患者,并确定了氢氯噻嗪试验诊断 GS 的界值(△FE Cl ≤2.86%)。以基因诊断为金标准,氢氯噻嗪试验诊断 GS 的灵敏度和特异度分别为 93.1%和 100%,优于低血镁和低尿钙等传统诊断方法[22] 。氢氯噻嗪试验是相对安全的诊断方法,目前尚无相关低血压、严重低钾血症等不良事件报道[22,35-37] ,可用于儿童患者 [35,37-38] ,但试验过程中应注意观察血压、血钾水平。与基因检测相比,氢氯噻嗪试验具有简便易行、价格低廉等特点,适用于各级医院,特别是基层医院在鉴别失盐性肾病肾小管受累部位及程度方面。
2.3 临床分级与分型
推荐意见:
- (6)GS 临床表现多样,临床症状严重程度与低钾、低镁血症程度相关,但个体间存在差异(证据等级:3;推荐强度:B)。
- (7)部分 GS 患者血镁正常,正常血镁较低血镁患者临床表现轻,可将血镁作为临床分型的依据之一(证据等级:3;推荐强度:B)。
- (8)GS 患者的尿前列腺素 E2(prostaglandin E2,PGE2)代谢产物水平可升高,且与临床表现严重程度相关,可将其作为临床分型指导治疗的依据之一(证据等级:3;推荐强度:B)。
2.3.1 根据临床表现严重程度分级
2016 年 KDIGO 根据 WHO 不良事件分级,将低钾血症和低镁血症分为 4 级,用以评价GS因电解质紊乱所带来的临床风险(https://kdigo.org/conferences/gitelman/),见表 4。
| 分级 | 1级 | 2级 | 3级 | 4级 |
|---|---|---|---|---|
| 血钾(mmol/L) | 3.0~3.4 | 2.5~2.9 | 2.0~2.4 或强效替代治疗,或 住院治疗 |
<2.0 或低血钾伴轻瘫、肠梗阻,或危及生命的心律失常 |
| 血镁(mmol/L) | 0.6~0.7 | 0.45~0.59 | 0.3~0.44 | <0.30 或伴危及生命的心律失常或手足抽搐 |
实际工作中,在相同血钾/血镁水平下,因患者对血钾/血镁的耐受程度不同,可出现不同的临床表现。参照美国国立卫生研究院 2017 年发布的常见不良事件评价标准(commonterminology criteria for adverse events,CTCAE)5.0 版本(https://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm#ctc_60),结合临床实际情况,根据 GS 严重程度将其临床表现分为 4 级(表 5)。用于分级的主要依据为:一般症状、泌尿系统症状、神经肌肉系统症状、消化系统症状、血电解质水平、血气分析结果和心电图 QT 间期等。
表 5 Gitelman 综合征临床表现分级建议
| 级别 | 临床表现 |
|---|---|
| A级 | 无症状低钾血症(偶然化验发现低钾血症);或仅轻微乏力、疲劳、多饮、多尿等,不影响日常生活,不对患者造成困扰 |
| B级 | 自觉症状明显,一定程度上影响日常生活质量 |
| C级 | 严重或具有重要医学意义,但不会危及生命;导致住院或住院时间延长;严重影响日常生活,可能影响自理能力 |
| D级 | 可能危及生命,需紧急治疗 |
2.3.2 根据性别分型
多数研究认为,男性 GS 患者较女性临床表型更重[6,7,31,39-41] 。研究显示,男性患者 18 岁前的发病比例较女性更高,血钾更低、尿钠及尿氯排泄更多,多尿症状更为突出[6] 。Tseng等[7] 的队列研究显示,男性患者的低血钾程度更重、血浆醛固酮水平更低。但国外亦有研究发现女性患者的尿钾排泄率更高、补钾量更大[42] ,另有研究未观察到症状和生活质量的性别差异[43] 。
2.3.3 根据血镁水平分型
根据国内文献报道,正常血镁的 GS 患者比率约为 8%~22%[7,16-18] ,部分正常血镁的患者可进展为低镁血症[44] 。研究指出正常血镁的 GS 患者电解质紊乱程度、NCC 功能损害程度均较低血镁的 GS 患者轻[45] 。在肾脏组织观察到远端小管镁离子转运蛋白(transient receptor potential channel melastatin subtype 6,TRPM6) 与 NCC 共表达,正常血镁患者的TRPM6表达与健康人接近,而低血镁患者的 TRPM6 表达显著降低,从而为正常血镁亚型的确立提供了病理生理学基础[45] 。目前认为血镁水平有助于判断 GS 患者的病情严重程度,可将血镁作为临床分型的依据之一。
2.3.4 根据尿前列腺素代谢产物分型
研究证实,环氧化酶(cyclooxygenase,COX)抑制剂可改善 GS 顽固性低钾、低镁血症及相关临床症状[46-49] 。此外,研究观察到 GS 患者血浆和尿液前列腺素 E2 代谢产物(prostaglandin E2 metabolites,PGEM)水平均较健康人群升高,高尿GEM 组患者出现夜尿增多和呼吸困难的比率更高,代谢性碱中毒更严重、尿钾和尿氯排泄量更高,对氢氯噻嗪的反应比低尿PGEM 组差[50] 。因此,尿 PGEM 水平可作为 GS 的分型依据,有助于识别 GS 高危患者和筛选COX 抑制剂治疗的合适人群。目前,临床上可采用 PGEM EIA 试剂盒测定尿 PGEM含量。
3 治疗策略
推荐意见:
- (9)GS 患者可通过饮食和药物补充钾和镁。补钾建议采用氯化钾,补镁建议采用有机酸盐制剂;严重低钾血症或低镁血症者可予静脉补充(证据等级:3;推荐强度:B)。
- (10)当 GS 患者存在顽固性电解质紊乱及相关临床症状,或依赖大剂量和/或静脉补钾补镁治疗,或补钾补镁不耐受时,推荐联合使用保钾利尿剂(如螺内酯、依普利酮和阿米洛利)、COX 抑制剂(如吲哚美辛)(证据等级:1b;推荐强度:A),也可联合使用 ACEI/ARB 类药物,但需监测血压(证据等级:3;推荐强度:B)。
GS 的主要治疗目标为改善患者症状并提高生活质量。治疗方法主要包括终身电解质替代治疗和基于发病机制的治疗,需长期规律随访与监测。
3.1 电解质替代治疗
3.1.1 饮食调整
首先建议饮食补充。多食用富含钾、镁的食物,如谷物、薯类、牛奶、蔬菜、坚果、黑巧克力等。如未合并高血压,鼓励多进食含盐饮食。
3.1.2 药物替代治疗
3.1.2.1 补钾药物
建议补充氯化钾,可同时补充尿液中丢失的氯离子,且不会加重代谢性碱中毒。当患者无法耐受口服补钾或有严重低钾血症时(如出现心律失常、软瘫、呼吸衰竭、横纹肌溶解等并发症时),可予静脉补钾治疗。通常认为 GS 患者的血钾纠正目标为 3.0 mmol/L 以上[1] 。
3.1.2.2 补镁药物
合并低镁血症时,应优先补镁治疗,可减少尿钾排泄,有助于低钾血症的纠正[51-52] 。同时有助于改善患者神经肌肉症状和抑郁状态[53] ,减轻生长发育迟滞及钙质沉着等 [1,54] 。补镁药物剂型方面,可选用有机酸盐制剂(如门冬氨酸盐、枸橼酸盐、乳酸盐等)口服,生物利用度更高,可分次随餐服用以减轻消化道症状。当出现严重低镁血症伴手足抽搐或心律失常时,可静脉补镁治疗。通常认为 GS 患者合适的血镁水平为 0.6 mmol/L 以上[1] 。
3.2 基于发病机制的治疗
当GS患者存在顽固性电解质紊乱及相关临床症状,或依赖大剂量和/或静脉补钾补镁治疗,或补钾补镁治疗不耐受时,可予联合用药以改善电解质紊乱、减少替代治疗药物剂量。联合用药主要包括 3 大类:保钾利尿剂、COX 抑制剂、血管紧张素转化酶抑制剂(angiotensin converting enzyme inhibitor,ACEI)/血管紧张素受体拮抗剂(angiotensin receptor blocker,ARB)。目前,关于 GS 联合用药的文献仅包含 1 项随机病例交叉研究[49] 和 1 项回顾性病例对照研究[16] ,其余多为病例报道,有待更多临床研究进一步证实。
3.2.1 保钾利尿剂
主要包括醛固酮拮抗剂类药物(如螺内酯、依普利酮)和非醛固酮拮抗剂类药物(如阿米洛利)。醛固酮拮抗剂类药物通过拮抗醛固酮对远曲小管和集合管多种离子通道的作用,降低尿钾排泄,提高血钾水平。其中螺内酯具有抗雄激素活性,可出现男性乳房发育等副作用;而依普利酮为选择性醛固酮受体拮抗剂,对雄激素和孕激素受体亲和力极低,无螺内酯的抗雄激素相关副作用。非醛固酮拮抗剂类药物通过直接拮抗上皮钠通道,减少钾的排泄。1 项随机病例交叉研究发现,采用依普利酮 150 mg/d、阿米洛利 20 mg/d 可使 GS 患者的血钾水平分别提高 0.15 mmol/L、0.19 mmol/L[49] 。目前,尚无研究比较螺内酯与依普利酮的疗效差别,从性激素相关副作用考虑,依普利酮因副作用较小而更具优势。
3.2.2 环氧化酶抑制剂
鉴于部分 GS 患者尿 PGE2 及 PGEM 水平增高[46,50] ,且尿 PGEM 水平高的患者临床表现更重[50] ,COX 抑制剂(如吲哚美辛)有助于部分 GS 患者改善低钾血症及相关症状,通过测定尿PGEM 水平有助于筛选此类患者。1 项随机病例交叉研究显示,使用吲哚美辛 75 mg/d 可使 GS患者的血钾水平升高 0.38 mmol/L[49] 。但需注意药物副作用,特别是肾小球滤过率降低和消化性溃疡等。
3.2.3 血管紧张素转化酶抑制剂 / 血管紧张素受体拮抗剂
ACEI/ARB 主要通过抑制 GS 患者激活的肾素-血管紧张素系统(renin angiotensin system,RAS)发挥作用,用药时须注意监测血压和肾功能水平[1-2] 。
3.3 随访监测和预后
GS 患者的管理强调个体化。患者应每 3~6 个月到肾内科和内分泌科随诊 1 次,评估相关症状、肾功能和电解质水平、并发症情况(心律失常、糖和骨代谢异常等)、心理情绪状态、药物副作用等,调整药物治疗方案和剂量,疏导可能发生的情绪障碍。如 GS 患者合并有其他疾病,用药时应慎用易诱发血钾或血镁降低的药物,如β2 受体激动剂[55] 、质子泵抑制剂[56] 、高剂量胰岛素和葡萄糖、非保钾利尿剂、泻药等。多数 GS 患者预后良好,但亦有引起严重表型、甚至危及生命的个案报道,因此早期诊断、规范化治疗和管理对改善预后具有积极意义。
4 慢性并发症及合并症的管理
推荐意见:
糖代谢异常
- (11)GS 患者可出现糖代谢异常,包括空腹血糖受损、糖耐量减低和 2 型糖尿病,应定期监测葡萄糖耐量和血糖水平(证据等级:3;推荐强度:B)。
- (12)补钾补镁及螺内酯治疗可改善 GS 患者的糖代谢异常(证据等级:3;推荐强度:B)。
骨关节改变
- (13)儿童起病的 GS 患者可出现身材矮小,补钾补镁治疗可改善部分患者的身高增长速度;如常规治疗后患者身高增长不理想,可行生长激素刺激试验,对于生长激素缺乏者可予生长激素治疗(证据等级:3;推荐强度:B)。
- (14)补镁治疗可预防或减少焦磷酸钙沉积病(calcium pyrophosphate deposition disease,CPPD);发作性关节肿痛急性期可予非甾体抗炎药(nonsteroidal anti-inflammatory drugs,NSAIDs)或小剂量秋水仙碱对症治疗(证据等级:3;推荐强度:B)。
4.1 糖代谢
研究显示,14%~60% 的 GS 患者可出现糖代谢异常[17,57-59] 。Yuan 等 [57] 研究发现,53.6%的GS 患者存在糖代谢异常,包括空腹血糖受损(3.6%)、糖耐量减低(28.6%)和 2 型糖尿病(21.4%),且存在胰岛素抵抗。其发生机制为长期的低钾血症导致胰岛素分泌减少,低镁血症损害胰岛素信号传导途径、降低胰岛素对葡萄糖的敏感性[57,60] ,以及继发性醛固酮增多引起的胰岛素抵抗。定期监测葡萄糖耐量和血糖有助于早期诊断,补钾补镁及螺内酯治疗可改善葡萄糖代谢受损[7] 。
4.2 生长发育与骨关节改变
儿童时期发病的 GS 患者可出现身材矮小、生长迟缓[61] 。补钾补镁治疗可改善部分患儿的生长情况[31] ,如常规治疗后身高增长不理想,绝对身高在同年龄、同性别正常儿童身高均值-2SD 以下,生理年龄≥2 岁且≤14 岁,女孩骨龄≤10 岁或男孩骨龄≤11 岁,青春发育期前儿童(Tanner Ⅰ期),可行生长激素刺激试验,对于生长激素缺乏者可予生长激素治疗。GS 患者通常骨密度增加[62-63] ,因 NCC 失活可直接促使成骨细胞分化和骨矿盐沉积增加。此外,GS 患者可出现 CPPD,多在 50 岁之前,但很少出现慢性变形性关节炎[64-65] ,低镁血症是其重要原因 [65-66] 。补充镁剂、维持正常血镁可预防或减少 CPPD,发作性关节肿痛急性期可采用 NSAIDs 或小剂量秋水仙碱对症治疗[65] 。
4.3 其他肾小管疾病
GS 患者可出现近端小管磷酸盐重吸收减少[67] ,还可合并不完全性 Fanconi 综合征,经补钾治疗后,代谢性酸中毒、低钠血症和低磷血症均可改善,提示其近端小管功能障碍可继发于严重的低钾血症[68] 。
4.4 合并高血压
世界各国均有 GS 患者合并高血压的报道,高血压患者占比为 5%~44%[16,42,69] 。因人群高血压的发病率高,GS 患者出现高血压考虑为临床合并的可能性较大,但也不除外与 GS 患者的 RAS 持续激活、存在胰岛素抵抗有关[42,69] 。因此,高血压不应作为排除 GS 诊断的必要条件。
4.5 合并其他疾病
GS 患者最常合并的疾病为甲状腺疾病,包括 Grave’s 病、桥本氏甲状腺炎和甲状腺功能异常等,原因尚不明确[70] 。此外,GS 还可合并原发性醛固酮增多症 [71] 、干燥综合征 [72] 等可导致低钾血症的疾病,临床上需注意鉴别。
5 特殊情况管理
推荐意见:
合并妊娠
- (15)妊娠可加重低钾、低镁血症,特别是妊娠早期,应严格监测血电解质水平,并增加钾和镁的补充;分娩后血钾、血镁水平可改善(证据等级:3;推荐强度:B)。
- (16)可根据患者情况采用剖宫产或阴道分娩,围产期应多学科团队协作,进行密切监测(证据等级:3;推荐强度:B)。
围术期管理
- (17)因 GS 患者存在电解质、酸碱平衡紊乱,以及 QT 间期延长甚至恶性心律失常的风险,围术期应充分评估,强调多学科团队协作,制订个体化管理方案(证据等级:3;推荐强度:B)。
5.1 合并妊娠
GS 患者总体妊娠结局良好。妊娠相关并发症包括羊水过少、妊娠糖尿病、妊娠高血压、HELLP 综合征、胎盘早剥,胎儿死亡、自然流产、早产和宫内生长迟缓等[6] 。妊娠期低钾血症的严重并发症表现为室性心律失常、进行性肌无力、呼吸肌麻痹,甚至母婴死亡[73-74] 。目前文献多为个案报道,尚无法比较与健康人群的并发症发生率差异。
妊娠可加重低钾、低镁血症,主要发生于妊娠早期,因此应严格监测电解质水平,并增加钾和镁的补充;同时,需考虑妊娠期血容量增加产生的影响,酌情调整药物剂量;分娩后血钾、血镁水平可逐渐改善。由于 ACEI/ARB 类药物存在致畸风险,妊娠期禁用[1-2] 。尽管有妊娠期使用螺内酯未致新生儿性发育异常的报道,但普遍认为妊娠期应禁用[75-76] 。避免长期使用 NSAIDs,有导致胎儿动脉导管过早关闭的风险[75-76] 。GS 患者的分娩方式视具体情况而定,文献报道剖宫产的比率约为 50%[6] 。围产期密切监测病情和多学科团队协作非常重要,建议肾内科、营养科、产科、麻醉科和儿科医生共同参与患者的管理。
5.2 围术期管理
GS 患者的手术分级涵盖一至四级,麻醉方式多为全身麻醉,剖宫产采用椎管内麻醉,总体麻醉及手术过程顺利。术前应评估患者的电解质及酸碱平衡紊乱程度、心律失常等严重并发症的发生风险,密切监测血钾、血镁、心电图等指标,强调多学科团队协作、制订个体化管理方案。目前,关于 GS 患者围术期安全的血钾、血镁水平尚不明确,根据英国国家健康与临床卓越研究所的指南推荐,血钾应不低于 3.0 mmol/L,血镁应不低于 0.5 mmol/L[1,2,77] 。麻醉期间需注意低钾、低镁血症对麻醉药物药效的影响,以及麻醉药物对肾脏损伤的潜在风险。
6 小结
GS 是一种遗传性肾小管疾病,随着对疾病认识的深入,越来越多的证据显示 GS 并非简单的良性肾小管间质疾病,除电解质紊乱外,还可出现室性心律失常、横纹肌溶解、肾功能衰竭、生长发育迟缓等严重情况。近年来,国内 GS 相关研究取得长足发展,制订新的 GS 共识势在必行。因此,本文基于最新循证医学证据,从 GS 的临床表现与分型到规范化诊断、治疗和管理等方面达成了共识,有助于临床医师制订合理、准确的诊疗策略。未来,随着更多 GS 高质量临床研究证据的出现,共识将不断更新和完善。
作者贡献:
张抒扬牵头制订共识框架,并组建共识制订工作组;陈丽萌组织起草共识初稿;魏珉、刘晓清、刘雅萍、夏维波、刘俊涛、李雪梅对共识内容进行修订和审校。
利益冲突:
专家组 成员 (按姓氏首字母排序) :
陈丽萌(北京协和医院肾内科)
陈崴(中山大学附属第一医院肾内科)
程虹(首都医科大学附属北京安贞医院肾内科)
韩飞(浙江大学医学院附属第一医院肾脏病中心)
郝传明(复旦大学附属华山医院肾内科)
胡颖(浙江大学医学院附属第二医院肾内科)
李明喜(北京协和医院肾内科)
李雪梅(北京协和医院肾内科)
李月红(北京清华长庚医院肾内科)
刘俊涛(北京协和医院妇产科)
刘小荣(首都医科大学附属北京儿童医院肾内科)
刘晓清(北京协和医院感染内科 临床流行病学教研室)
刘雅萍(中国医学科学院基础医学研究所 医学遗传学系)
陆晨(新疆维吾尔自治区人民医院肾内科)
毛永辉(北京医院肾内科)
梅长林(上海长征医院肾内科)
秦岩(北京协和医院肾内科)
邱玲(北京协和医院检验科)
任红(上海交通大学医学院附属瑞金医院肾内科)
童安莉(北京协和医院内分泌科)
王悦(北京大学第三医院肾内科)
魏珉(北京协和医院儿科)
夏维波(北京协和医院内分泌科)
杨琼琼(中山大学附属第一医院肾内科)
姚丽(中国医科大学附属第一医院肾内科)
易杰(北京协和医院麻醉科)
张宏(北京大学第一医院肾内科)
张抒扬(北京协和医院疑难重症及罕见病国家重点实验室)
郑智华(中山大学附属第一医院肾内科)
秘书组成员 :
张磊(北京协和医院肾内科)
简珊(北京协和医院儿科)
牟利军(浙江大学医学院附属第二医院肾内科)
袁涛(北京协和医院内分泌科)
执笔者:
陈丽萌(北京协和医院肾内科)
张磊(北京协和医院肾内科)
简珊(北京协和医院儿科)
牟利军(浙江大学医学院附属第二医院肾内科)
袁涛(北京协和医院内分泌科)
彭晓艳(首都儿科研究所附属儿童医院肾内科)
蒋兰萍(中山大学附属第一医院肾内科)
赵冰彬(北京协和医院肾内科)
马亦昕(北京协和医院肾内科)
施潇潇(北京协和医院肾内科)
纪培丽(北京协和医院肾内科)
参考文献
- [1] Blanchard A,Bockenhauer D,Bolignano D,et al.Gitelman syndrome:consensus and guidance from a Kidney Disease:Improving Global Outcomes (KDIGO) Controversies Conference[J].Kidney Int,2017,91:24-33.
- [2] Gitelman 综合征诊治专家共识协作组.Gitelman 综合征诊治专家共识[J].中华内科杂志,2017,56:712-716.
- [3] Graham AJ, Gelfand G, McFadden SD,et al.Levels of evidence and grades of recommendations in general thoracic surgery[J].Can J Surg,2004,47:461-465.
- [4] 中华医学会内科学分会,王建祥,张奉春,等.中国成人血小板减少症诊疗专家共识[J].中华内科杂志,2020,59:498-510.
- [5] Jones J,Hunter D.Consensus methods for medical and health services research[J].BMJ,1995,311:376-380.
- [6] Zhang L,Peng X,Zhao B,et al.Clinical and laboratory features of female Gitelman syndrome and the pregnancy outcomes in a Chinese cohort[J].Nephrology (Carlton),2020,25:749-757.
- [7] Tseng MH,Yang SS,Hsu YJ,et al.Genotype,phenotype,and follow-up in Taiwanese patients with salt-losing tubulopathy associated with SLC12A3 mutation[J].J Clin Endocrinol Metab,2012,97:E1478-E1482.
- [8]Bulucu F,Vural A, Yenicesu M,et al.Association of Gitelman’s syndrome and focal lomerulosclerosis[J].Nephron,1998,79:244.
- [9]Rosado Rubio C,Fraile Gómez P,Gómez Muñoz MA,et al.C1q nephropathy in a patient with Gitelman syndrome[J].NDT Plus,2011,4:392-393.
- [10]Hanevold C,Mian A,Dalton R.C1q nephropathy in association with Gitelman syndrome:a case report[J]. Pediatr Nephrol,2006,21:1904-1908.
- [11] Chen Q,Wu Y,Zhao J,et al.A case of hypokalemia and proteinuria with a new mutation in the SLC12A3 Gene[J].BMC Nephrol,2018,19:275.
- [12] Konrad M,Nijenhuis T,Ariceta G,et al.Diagnosis and management of Bartter syndrome:executive summary of the consensus and recommendations from the European Rare Kidney Disease Reference Network Working Group for Tubular Disorders[J].Kidney Int,2021,99:324-335.
- [13]Shao L,Ren H,Wang W,et al.Novel SLC12A3 mutations in Chinese patients with Gitelman’s syndrome[J].Nephron Physiol,2008,108:29-36.
- [14]Qin L,Shao L,Ren H,et al.Identification of five novel variants in the thiazide-sensitive NaCl co-transporter gene in Chinese patients with Gitelman syndrome[J].Nephrology (Carlton),2009,14:52-58.
- [15] Miao M , Zhao CQ , Wang XL , et al.Clinical and genetic analyses of Chinese patients with Gitelman syndrome[J].Genet Mol Res,2016.doi:10.4238/gmr.15027859.
- [16]Wang F,Shi C,Cui Y,et al.Mutation profile and treatment of Gitelman syndrome in Chinese patients[J].Clin Exp Nephrol,2017,21:293-299.
- [17] Liu T,Wang C,Lu J,et al.Genotype/phenotype analysis in 67 Chinese patients with Gitelman’s syndrome[J].Am J Nephrol,2016,44:159-168.
- [18]Ma J,Ren H,Lin L,et al.Genetic features of Chinese patients with Gitelman syndrome:sixteen novel SLC12A3 mutations identified in a new cohort[J].Am J Nephrol,2016,44:113-121.
- [19] Zeng Y,Li P,Fang S,et al.Genetic analysis of SLC12A3 gene in Chinese patients with Gitelman syndrome[J].Med Sci Monit,2019,25:5942-5952.
- [20]Vargas-Poussou R,Dahan K,Kahila D,et al.Spectrum of mutations in Gitelman syndrome[J].J Am Soc Nephrol,2011,22:693-703.
- [21]Glaudemans B,Yntema HG,San-Cristobal P,et al.Novel NCC mutants and functional analysis in a new cohort of patients with Gitelman syndrome[J].Eur J Hum Genet,2012,20:263-270.
- [22]Peng X,Zhao B,Zhang L,et al.Hydrochlorothiazide test as a tool in the diagnosis of Gitelman syndrome in Chinese patients[J].Front Endocrinol (Lausanne),2018,9:559.
- [23] Nozu K,Iijima K,Nozu Y,et al.A deep intronic mutation in the SLC12A3 gene leads to Gitelman syndrome[J].Pediatr Res,2009,66:590-593.
- [24] Nozu K,Nozu Y,Nakanishi K,et al.Cryptic exon activation in SLC12A3 in Gitelman syndrome[J].J Hum Genet,2017,62:335-337.
- [25]Lo YF,Nozu K,Iijima K,et al.Recurrent deep intronic mutations in the SLC12A3 gene responsible for Gitelman’s syndrome[J].Clin J Am Soc Nephrol,2011,6:630-639.
- [26] Nagano C,Nozu K,Morisada N,et al.Detection of copy number variations by pair analysis using next-generation sequencing data in inherited kidney diseases[J].Clin Exp Nephrol,2018,22:881-888.
- [27] Tavira B,Gómez J,Santos F,et al.A labor- and cost-effective non-optical semiconductor (Ion Torrent) next-generation sequencing of the SLC12A3 and CLCNKA/B genes in Gitelman’s syndrome patients[J].J Hum Genet,2014,59:376-380.
- [28]Ashton EJ,Legrand A,Benoit V,et al.Simultaneous sequencing of 37 genes identified causative mutations in the majority of children with renal tubulopathies[J].Kidney Int,2018,93:961-967.
- [29]Hureaux M,Ashton E,Dahan K,et al.High-throughput sequencing contributes to the diagnosis of tubulopathies and familial hypercalcemia hypocalciuria in adults[J].Kidney Int,2019,96:1408-1416.
- [30]Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J].Genet Med,2015,17:405-424.
- [31]Riveira-Munoz E,Chang Q,Godefroid N,et al.Transcriptional and functional analyses of SLC12A3 mutations:new clues for the pathogenesis of Gitelman syndrome[J].J Am Soc Nephrol,2007,18:1271-1283.
- [32]De Jong JC,Van Der Vliet WA,Van Den Heuvel LP,et al.Functional expression of mutations in the human NaCl cotransporter:evidence for impaired routing mechanisms in Gitelman’s syndrome[J].J Am Soc Nephrol,2002,13:1442-1448.
- [33]Kunchaparty S,Palcso M,Berkman J,et al.Defective processing and expression of thiazide-sensitive Na-Cl cotransporter as a cause of Gitelman’s syndrome[J].Am J Physiol,1999,277:F643-F649.
- [34] Sabath E,Meade P,Berkman J,et al.Pathophysiology of functional mutations of the thiazide-sensitive Na-Cl cotransporter in Gitelman disease[J].Am J Physiol Renal Physiol,2004,287:F195-F203.
- [35] Colussi G,Bettinelli A,Tedeschi S,et al.A thiazide test for the diagnosis of renal tubular hypokalemic disorders[J].Clin J Am Soc Nephrol,2007,2:454-460.
- [36]彭晓艳,蒋兰萍,袁涛,等.氯离子清除试验在 Gitelman 综合征鉴别诊断中的应用[J].中国医学科学院学报,2016,38:275-282.
- [37] Nozu K,Iijima K,Kanda K,et al.The pharmacological characteristics of molecular-based inherited salt-losing tubulopathies[J].J Clin Endocrinol Metab,2010,95:E511-E518.
- [38] 张军,陈秋莉,李燕虹,等.呋塞米/氢氯噻嗪负荷试验在儿童 Bartter 综合征和 Gitelman 综合征鉴别诊断中的应用[J].临床儿科杂志,2016,34:891-893.
- [39] Lin SH,Cheng NL,Hsu YJ,et al.Intrafamilial phenotype variability in patients with Gitelman syndrome having the same mutations in their thiazide-sensitive sodium /chloride cotransporter[J].Am J Kidney Dis,2004,43:304-312.
- [40]Fukuyama S,Okudaira S,Yamazato S,et al.Analysis of renal tubular electrolyte transporter genes in seven patients with hypokalemic metabolic alkalosis[J].Kidney Int,2003,64:808-816.
- [41]Lin SH,Shiang JC,Huang CC,et al.Phenotype and genotype analysis in Chinese patients with Gitelman’s syndrome[J].J Clin Endocrinol Metab,2005,90:2500-2507.
- [42] Berry MR,Robinson C,Karet Frankl FE.Unexpected clinical sequelae of Gitelman syndrome:hypertension in adulthood is common and females have higher potassium requirements[J].Nephrol Dial Transplant,2013,28:1533-1542.
- [43] Cruz DN,Shaer AJ,Bia MJ,et al.Gitelman’s syndrome revisited:an evaluation of symptoms and health-related quality of life[J].Kidney Int,2001,59:710-717.
- [44] Tammaro F,Bettinelli A,Cattarelli D,et al.Early appearance of hypokalemia in Gitelman syndrome[J].Pediatr Nephrol,2010,25:2179-2182.
- [45]Jiang L,Chen C,Yuan T,et al.Clinical severity of Gitelman syndrome determined by serum magnesium[J].Am J Nephrol,2014,39:357-366.
- [46]Larkins N,Wallis M,McGillivray B,et al.A severe phenotype of Gitelman syndrome with increased prostaglandin excretion and favorable response to indomethacin[J].Clin Kidney J,2014,7:306-310.
- [47]Jeck N,Schlingmann KP,Reinalter SC,et al.Salt handling in the distal nephron:lessons learned from inherited human disorders[J].Am J Physiol Regul Integr Comp Physiol,2005,288:R782-R795.
- [48]Liaw LC,Banerjee K,Coulthard MG.Dose related growth response to indometacin in Gitelman syndrome[J].Arch Dis Child,1999,81:508-510.
- [49]Blanchard A,Vargas-Poussou R,Vallet M,et al.Indomethacin,amiloride,or eplerenone for treating hypokalemia in Gitelman syndrome[J].J Am Soc Nephrol,2015,26:468-475.
- [50]Peng X,Jiang L,Chen C,et al.Increased urinary prostaglandin E2 metabolite:A potential therapeutic target of Gitelman syndrome[J].PLoS One,2017,12:e0180811.
- [51]Cho YJ,Park GT,Cho YJ,et al.Renal potassium wasting and hypocalciuria ameliorated with magnesium repletion in Gitelman’s syndrome[J].J Korean Med Sci,1997,12:157-159.
- [52] Robinson CM,Karet Frankl FE.Magnesium lactate in the treatment of Gitelman syndrome:patient-reported outcomes[J].Nephrol Dial Transplant,2017,32:508-512.
- [53] Hayashi M.Gitelman’s syndrome and hypomagnesemia[J].Intern Med,2004,43:351-352.
- [54] Ayuk J,Gittoes NJ.How should hypomagnesaemia be investigated and treated? [J] Clin Endocrinol (Oxf),2011,75:743-746.
- [55] Sundram V,Daly E,Waldron M.Gitelman syndrome:Case report of a toddler following salbutamol inhaler[J].Archives of Disease in Childhood,2019,104:A182.
- [56] Tursi A.Gitelman syndrome triggered by proton-pump inhibitor use[J].Dig Liver Dis,2019,51:911.
- [57]Yuan T,Jiang L,Chen C,et al.Glucose tolerance and insulin responsiveness in Gitelman syndrome patients[J].Endocr Connect,2017,6:243-252.
- [58] Blanchard A,Vallet M,Dubourg L,et al.Resistance to insulin in patients with Gitelman syndrome and a subtle intermediate phenotype in heterozygous carriers:a cross-sectional study[J].J Am Soc Nephrol,2019,30:1534-1545.
- [59]Ren H,Qin L,Wang W,et al.Abnormal glucose metabolism and insulin sensitivity in Chinese patients with Gitelman syndrome[J].Am J Nephrol,2013,37:152-157.
- [60] Knoers NV,Levtchenko EN.Gitelman syndrome[J].Orphanet J Rare Dis,2008,3:22.
- [61] Fujimura J,Nozu K,Yamamura T,et al.Clinical and Genetic Characteristics in Patients With Gitelman Syndrome[J].Kidney Int Rep,2018,4:119-125.
- [62] Cruz DN.The renal tubular Na-Cl co-transporter (NCCT):a potential genetic link between blood pressure and bone density?[J] Nephrol Dial Transplant,2001,16:691-694.
- [63] Nicolet-Barousse L,Blanchard A,Roux C,et al.Inactivation of the Na-Cl co-transporter (NCC) gene is associated with high BMD through both renal and bone mechanisms:analysis of patients with Gitelman syndrome and Ncc null mice[J].J Bone Miner Res,2005,20:799-808.
- [64] Gutierrez M,Silveri F,Bertolazzi C,et al.Gitelman syndrome,calcium pyrophosphate dihydrate deposition disease and crowned dens syndrome.A new association? [J] Rheumatology (Oxford),2010,49:610-613.
- [65] Favero M,Calò LA,Schiavon F,et al.Miscellaneous non-inflammatory musculoskeletal conditions.Bartter’s and Gitelman’s diseases[J].Best Pract Res Clin Rheumatol,2011,25:637-648.
- [66] Iqbal Z,Sayer JA.Chondrocalcinosis and Gitelman syndrome[J].QJM,2016,109:563-564.
- [67] Viganò C,Amoruso C,Barretta F,et al.Renal phosphate handling in Gitelman syndrome—the results of a case-control study[J].Pediatr Nephrol,2013,28:65-70.
- [68] Bouchireb K,Boyer O,Mansour-Hendili L,et al.Fanconi syndrome and severe polyuria:an uncommon clinicobiological presentation of a Gitelman syndrome[J].BMC Pediatr,2014,14:201.
- [69] Balavoine AS,Bataille P,Vanhille P,et al.Phenotype-genotype correlation and follow-up in adult patients with hypokalaemia of renal origin suggesting Gitelman syndrome[J].Eur J Endocrinol,2011,165:665-673.
- [70] Zhou H,Liang X,Qing Y,et al.Complicated Gitelman syndrome and autoimmune thyroid disease:a case report with a new homozygous mutation in the SLC12A3 gene and literature review[J].BMC Endocr Disord,2018,18:82.
- [71] Miao Z,Gao Y,Bindels RJ,et al.Coexistence of normotensive primary aldosteronism in two patients with Gitelman’s syndrome and novel thiazide-sensitive Na-Cl cotransporter mutations[J].Eur J Endocrinol,2009,161:275-283.
- [72]Mishima E,Mori T,Sohara E,et al.Inherited,not acquired,Gitelman syndrome in a patient with Sjögren’s syndrome:importance of genetic testing to distinguish the two forms[J].CEN Case Rep,2017,6:180-184.
- [73]Elkoundi A,Kartite N,Bensghir M,et al.Gitelman syndrome:a rare life-threatening case of hypokalemic paralysis mimicking Guillain-Barré syndrome during pregnancy and review of the literature[J].Clin Case Rep,2017,5:1597-1603.
- [74]Srinivas SK,Sukhan S,Elovitz MA.Nausea,emesis,and muscle weakness in a pregnant adolescent[J].Obstet Gynecol,2006,107:481-484.
- [75]Mascetti L,Bettinelli A,Simonetti GD,et al.Pregnancy in inherited hypokalemic salt-losing renal tubular disorder[J].Obstet Gynecol,2011,117:512-516.
- [76]Moustakakis MN,Bockorny M.Gitelman syndrome and pregnancy[J].Clin Kidney J,2012,5:552-555.
- [77] Gallagher H,Soar J,Tomson C.New guideline for perioperative management of people with inherited salt-wasting alkaloses[J].Br J Anaesth,2016,116:746-749.

若有收获,就点个赞吧
0 人点赞
