参考:生信技能树公众号、单细胞天地公众号、周运来就是我(简书)。 备注:仅作为笔记学习,无任何商业用途。
单细胞测序基础知识
单细胞测序
学习scRNA-seq先了解一下Bulk RNA-seq
- Bulk RNA-seq:测量一个大的细胞群体中每一个基因的平均表达水平,对比较转录组学(例如比较 不同物种的相同组织) 是有帮助的,但对于研究异质性系统(例如,复杂的组织如脑)还是不够的,对于基因表达的本质研究不够深入。
- scRNA-seq:
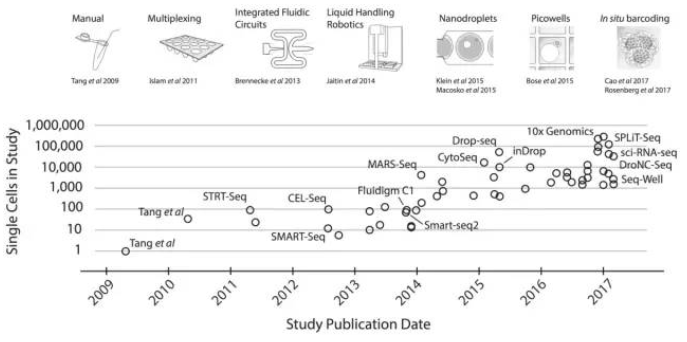
- 2009年由Tang首创,随着测序技术的发展及测序成本的的下降,2014年才逐渐流行开来。它测定的是细胞种群中每个基因的表达量分布,对于研究特定细胞转录组的变化是重要的。
- 通量:测定的细胞量也由最初的10个左右,达到了现在的百万并且还在不断递增,向着高通量的方向发展。
- 测序流程:现在主流的主要10X Genomics Chromium(较多细胞),SAMRT-seq2(较多基因)和Fluidigm C1等。当然还有其他的如:CELL-seq、Drop-seq、mas-seq和Wafergen ICELL8等。
单细胞测序流程
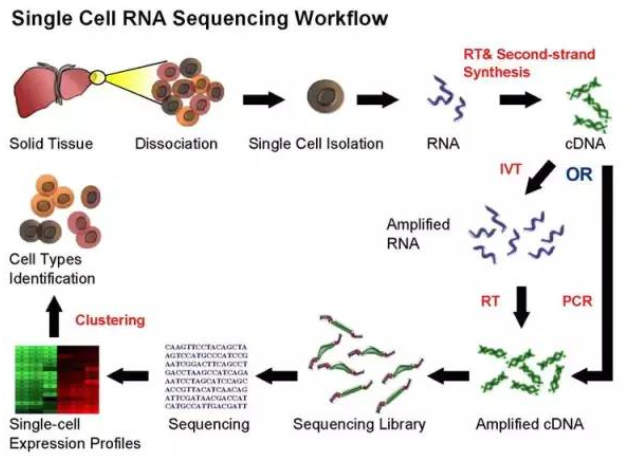
方法主要分为两大部分:定量与分离
- 单细胞定量包括两种类型:全长以及基于标签(tag)。前者对每个转录本都试图获得一致的read覆盖度,后者只捕获5’或者3’端的RNA。定量方法的选择也影响了后续分析的方法选择。理论上,全长的方法应该得到转录本的平均覆盖度,但是实际上,覆盖度经常是有偏差的。基于标签的方法能够利用特异性分子标记(Unique Molecular Identifiers, UMIs)提高定量准确度,但是呢,这种限制了转录组一端的方法有降低了转录本的可拼接性,让以后的isoform识别变得困难。
单细胞分离(Isolation of Single Cells)
单细胞分离方法:包括有限稀释法(Limiting dilution technique)显微操作法(micromanipulation)、流式细胞分选(FACS)、激光捕获显微分离(LCM)、微孔(microwell)、微流控(microfluidics)和微滴(droplet)。- 有限稀释技术(Limiting dilution technique)是利用移液管稀释分离细胞,这种方法的主要缺点是效率低下,成功率20%左右。
- 显微操作法(micromanipulation)是一种经典的方法,用于从少量细胞样本中提取细胞,如早期胚胎或未培养的微生物,缺点是耗时和低通量的。
- 流式细胞分选(FACS)广泛用于分离单个细胞,在悬浮状态下需要较大的起始体积(>10,000个细胞)。
- 激光捕获显微分离(LCM)是利用计算机辅助的激光系统将单个细胞从固体组织中分离出来。
- 微孔microwell 优势是可以与荧光触发细胞分离技术(FACS)结合,实现基于表面标记的细胞选择。对于想要分离特定细胞群的研究很有效;另外一个优势就是,可以对细胞进行拍照,帮助确定孔中是否存在损伤的或者重复的细胞。缺点是通量低,对每个细胞进行操作需要很大的工作量。
- 微流控(microfluidics)相对微孔,它的通量更高。缺点是只有10%的细胞能被捕获到,加入处理的细胞类型比较稀少,那么能被芯片捕获的就更少了,通量不高,成本高。
- 微滴技术 是将单个细胞包裹在µl级别的液滴中,液滴被搭载到建库所用的酶上,每个微滴包含一个独特的条码(barcode),由那个被包装好的细胞产生的所有reads都被贴上了该条码,也是为了之后对于不同细胞reads的分辨。一般微滴技术的通量最大,查资料得知一秒能包装7万个以上的液滴,并且建库费用合适—0.05美元/细胞。但是高额测序费用又成了他的短板,鉴定出的一般只有一千多个差异转录本,覆盖度还是比较低的
分析流程
黄色部分对于高通量数据的处理都是差不多的流程;
橙色的部分需要整合多个转录组分析流程以及显著性分析,来解决单细胞测序的技术误差;
蓝色是下游表达量、通路、互作网络等分析,需要使用针对单细胞研发的方法。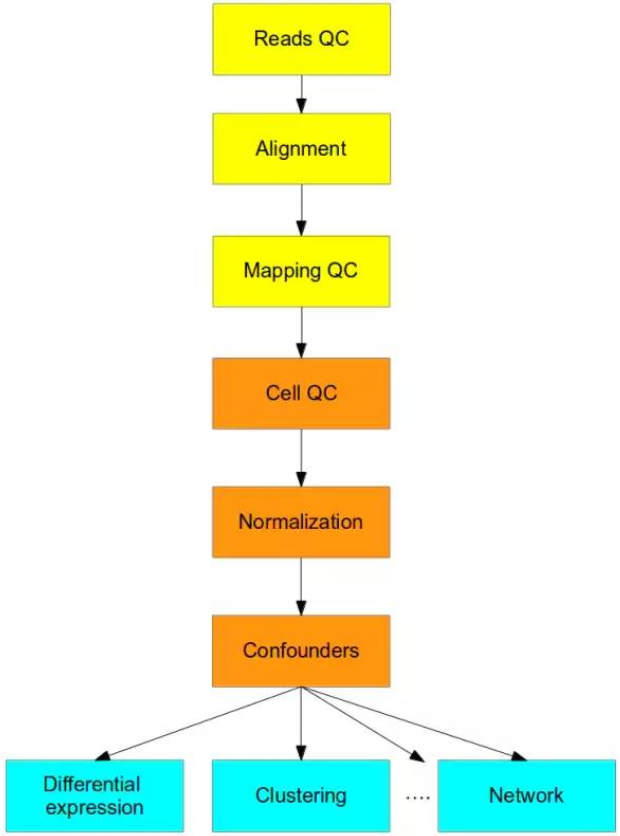
单细胞测序下游分析流程
很大概率上你并不会需要自己走上游流程,主要是因为对计算资源的消耗,实验室搭建上游流程成本太高,还不如一次性付费让公司做出来表达矩阵给到你后下游慢慢探索。过滤
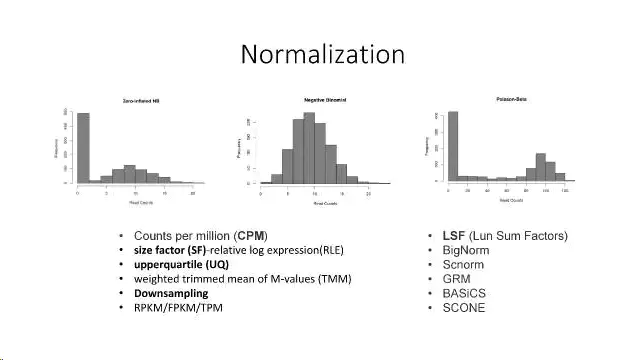
根据基因的表达量等特征,对细胞进行过滤,通常的做法就是指定一个阈值,比如要求一个细胞中检测到的基因数必须大于100,才可以进入到下游分析,如果小于这个数字,就过滤掉该细胞。需要强调的是,在设定过滤的阈值时,需要人为判断,这样的设定方式会受到主观因素的干扰,所以往往都会指定一个非常小的过滤范围,保证只过滤掉极少数的离群值点归一化
聚类分析
聚类分析用于识别细胞亚型(如细胞异质性、细胞分化周期的判定等),如在R包Seurat中,不是直接对所有细胞进行聚类分析,而是首先进行PCA主成分分析,然后挑选贡献量最大的几个主成分(也相当于做了特征选择),用挑选出的主成分的值来进行聚类分析。2019年的Nature Review上面发表了一篇文章来讨论单细胞测序数据聚类遇到的挑战,聚类分析是后续分析的起点,聚类方法的选择显得格外重要,同时也是对已有聚类算法的挑战。
常用的有图聚类和k-means聚类算法。
t-SNE是目前来说效果最好的数据降维与可视化方法,但是它的缺点也很明显,比如:占内存大,运行时间长。但是,当我们想要对高维数据进行分类,又不清楚这个数据集有没有很好的可分性(即同类之间间隔小,异类之间间隔大),可以通过t-SNE投影到2维或者3维的空间中观察一下。如果在低维空间中具有可分性,则数据是可分的;如果在高维空间中不具有可分性,可能是数据不可分,也可能仅仅是因为不能投影到低维空间。差异表达分析
细胞很多,每个细胞的基因也有很多,那么那些基因才是有意义的呢?需要一些统计手段来把这些基因识别出来,这就是差异表达分析,针对单细胞测序(特别是scRNA-seq)数据的特点,已经开发的算法和软件见下图: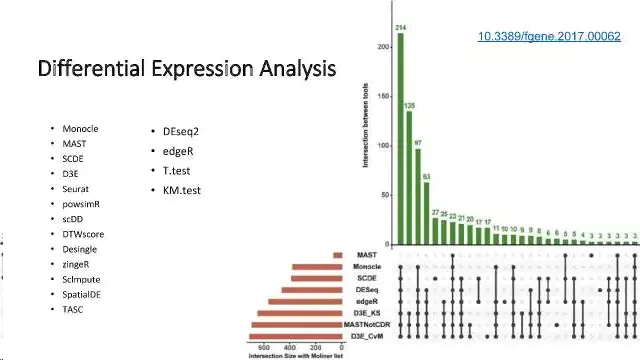
marker基因识别
通过差异分析来识别每个cluster下的标记基因,将该cluster下的细胞作为一组,其他cluster下的细胞作为另一组,然后进行差异分析。富集分析
一般提到富集分析,首先想到的就是GO、KEGG这两把刷子,然后还需要知道两个重要概念:前景基因、背景基因
前景基因:你关注的要重点研究的基因集;
背景基因:所有的基因集
比如做转录组测序,一般都要设置处理组和对照组,前景基因是处理和对照的差异基因,背景基因就是两组样本的全部表达基因。
另外还有一种是Gene Set Enrichment Analysis 基因集富集分析,用于评估一个基因集的基因在表型相关度排序中的分布趋势,进而判断它们对表型的贡献。蛋白互作网络分析
蛋白互作网络(protein protein interaction network,PPI network)分析有助于从系统的角度研究疾病分子机制、发现新药靶点等等。一个常用的PPI数据库是STRING数据库。STRING数据库是一个搜索已知蛋白质之间和预测蛋白质之间相互作用的数据库,该数据库可应用于2031个物种,包含960万种蛋白和1380万中蛋白质之间的相互作用。蛋白质之间的相互作用包括了直接的物理相互作用和间接的功能相关性。单细胞多组学分析
以上介绍的其实都是基于高通量的单细胞转录组分析思路,开创性的单细胞分析现在能够对基因组、表观基因组、转录组、蛋白质组和代谢组谱系进行分析。Cell旗下的Trends inBiotechnology早在2016年就综述了为同一的细胞提供复杂的谱系,将不同维度的分析组合成多组学分析的方法。单细胞多组学分析测量同一细胞内的细胞状态的不同方面的能力有望揭开细胞的基因组、表观基因、转录组、蛋白质组与代谢组之间的相关联系;可以揭示DNA甲基化、染色质于转录起始之间的复杂关系。同时这篇文章也给予单细胞多组学(单细胞系统生物学)极高的评价。一篇评论文章称:单细胞系统生物学是一个令人兴奋的新领域,关注单细胞作为生物学的核心将为基础科学提供见解,在生物技术和生物医学方法提供有效的应用机会。
新的分析点:
批次效应矫正
- RNA velocity分析
- 细胞间通信
- 分析百万单细胞的软件

若有收获,就点个赞吧
0 人点赞
