参考:生信技能树公众号、单细胞天地公众号、周运来就是我(简书)。 备注:仅作为笔记学习,无任何商业用途。
两种降维方式
需要标准化数据、寻找高变基因、PCA分析。
sce <- NormalizeData(sce, normalization.method = "LogNormalize", scale.factor = 10000)sce <- FindVariableFeatures(sce, selection.method = "vst", nfeatures = 2000)# 步骤 ScaleData 的耗时取决于电脑系统配置(保守估计大于一分钟)sce <- ScaleData(sce)sce <- RunPCA(object = sce, pc.genes = VariableFeatures(sce))
简单解释一下,这代码里面的FindVariableFeatures和RunPCA函数,是两种不同策略的降维。
- 首先FindVariableFeatures是硬过滤,根据一些统计指标,比如sd,mad,vst等等来判断你输入的单细胞表达矩阵里面的2万多个基因里面,最重要的2000个基因,其余的1.8万个基因下游分析就不考虑了。
- 然后RunPCA函数其实跑完之后2000个基因会转变为2000个维度,但是我们通常看前面的十几个维度就ok了,所以也是一个效率非常高的降维方式。
这个时候,你一定有疑问,为什么FindVariableFeatures是挑选2000个基因而不是其它数量呢?RunPCA函数跑完后我们应该是挑选前多少个维度呢?10个还是15个,还是20个还是50呢。
第一次降维,表达矩阵里面通常是2~3万的基因数量,需要筛选到千这个数量级,可以是1000-5000,都是可以
查看聚类分群
聚类分群是紧密连接的,细胞可以看做是空间的不同点,如果是二维平面空间,点与点之间的距离很方便计算,距离的远近就决定着细胞是否属于一个类群。
计算距离的公式很多,我们就不一一展开,但是需要注意的是二维平面空间,三维球体空间的细胞距离很方便计算,但是如果是50个维度的空间,计算几万个细胞之间的距离就很可怕了,如果是2000个维度,甚至是2万个维度,基本上个人计算机就可以放弃了。这就是为什么我们前面通常是需要降维的。
下面是一个典型的单细胞转录组项目数据处理的描述:

可以看到他们的第一步降维是,选取top 5000的表达量离散度大的基因,第二步降维是选取top20的主成分。使用KNN-graph的聚类,最终定下来了10个细胞亚群。
一般来说,如果单细胞转录组数据仅仅是文章生物学故事的一个环节,就会采取标准的seurat流程,如下所示:
如果你看的文献足够多,还会发现,在降维聚类分群之后,通常是有一个细胞在二维平面的散点图展示,如下所示,如果你足够心细,也会发现其实细胞的空间距离排布坐标通常是tSNE和umap来展现。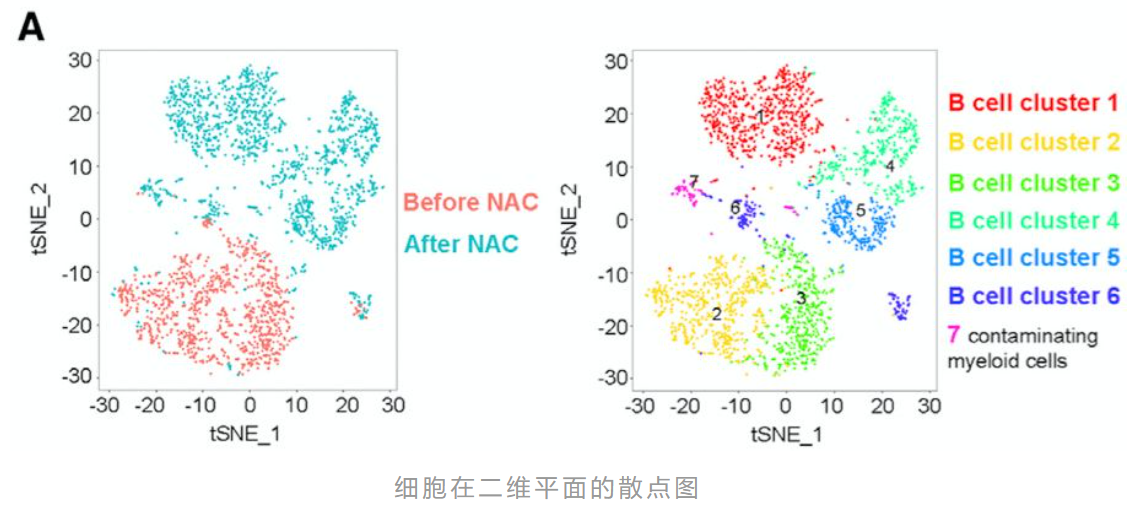
tSNE和UMAP
tSNE(t-distributed stochastic neighbor embedding) 是一种非线性降维的算法,是流形学习的一种,当然,你大概率上是无需理解流形学习的,认识一下其它流形学习的经典方法即可,包括:Isomap,LLE,LE和diffusion maps等。
- 困惑度(perplexity)可以表示细胞的邻近个数,在tSNE图上的直观反映是细胞点的分布是否紧凑。perplexity设置越大,细胞分布越紧凑。
- tSNE的参数设置:perplexity < (细胞数-1)/3,建议perplexity = 细胞数 / 50;
- tSNE倾向于保留数据的局部结构

若有收获,就点个赞吧
0 人点赞
