FastQC
FastQC软件可以对fastq格式的原始数据进行质量统计,评估测序结果,为下一步修剪过滤提供参考.
网址
https://www.bioinformatics.babraham.ac.uk/projects/fastqc/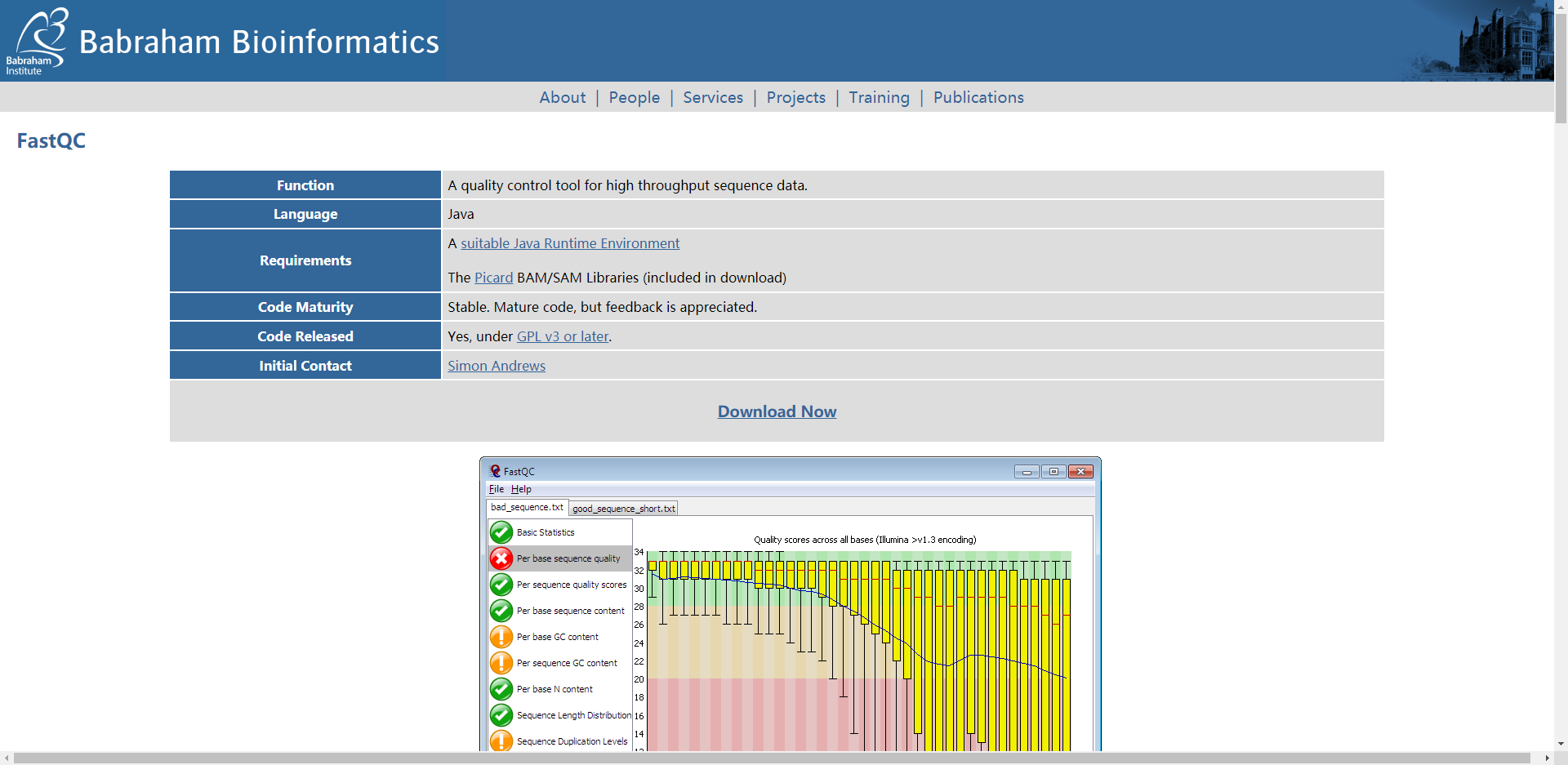
常见参数
-h/--help ## 打印文档信息-o/--outdir ## 输出目录(一定要存在,否则会报错)-j/--java ## 指定java路径,默认即可-f/--format ## 指定输入格式,默认为fastq。可以选择bam,sam-t/--threads ## 线程数-v/--versions ## 版本信息-d/--dir ## 质控过程中产生的临时文件
质量评估步骤
激活conda环境并进入工作目录
conda activate rna
准备原始数据文件
此步骤的数据应该来自下载数据,详见:
03 数据下载
使用FastQC软件对单个fastq文件评估
## 定义环境变量,分别是输出环境和原始文件环境qcdir=/home/data/vip13t21/project/GSE106901/data/qcfqdir=/home/data/vip13t21/project/GSE106901/data/rawData## fastqc进行质量评估fastqc -t 50 -o $qcdir $fqdir/SRR6292394.fastq.gz
批量数据评估
推荐使用vim命令写脚本,利用nohup和bash命令挂后台运行。
## 写脚本vim fastqc.sh## 脚本内容## 定义环境变量,分别是输出环境和原始文件环境qcdir=/home/data/vip13t21/project/GSE106901/data/qcfqdir=/home/data/vip13t21/project/GSE106901/data/rawData## fastqc进行质量评估fastqc -t 50 -o $qcdir $fqdir/SRR*.fastq.gz## 挂后台nohup bash fastqc.sh 1>fastqc.sh.log 2>&1 &

使用MultiQC命令整合FastQC结果
每个数据都会有一个质量检测文件,阅读不便,利用multiqc命令整合为一个文件。
评估数据解读

Basic Statistics

注:数据量统计方式

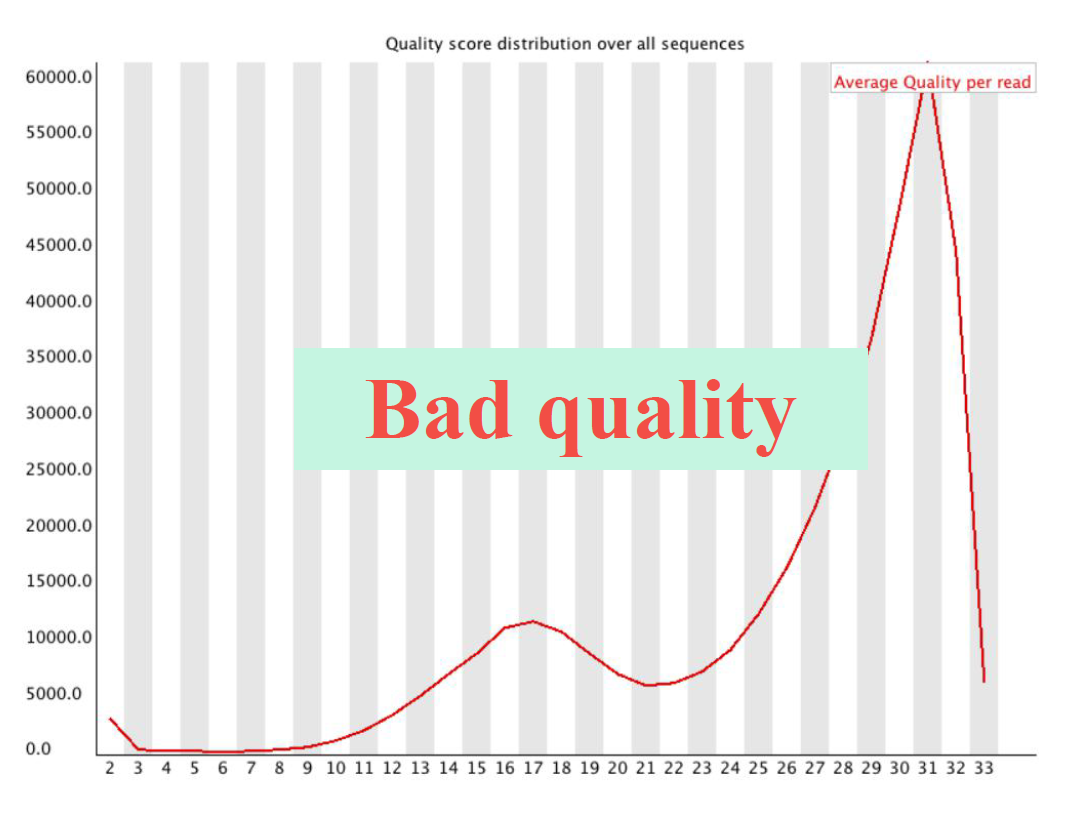
Per base sequence quality
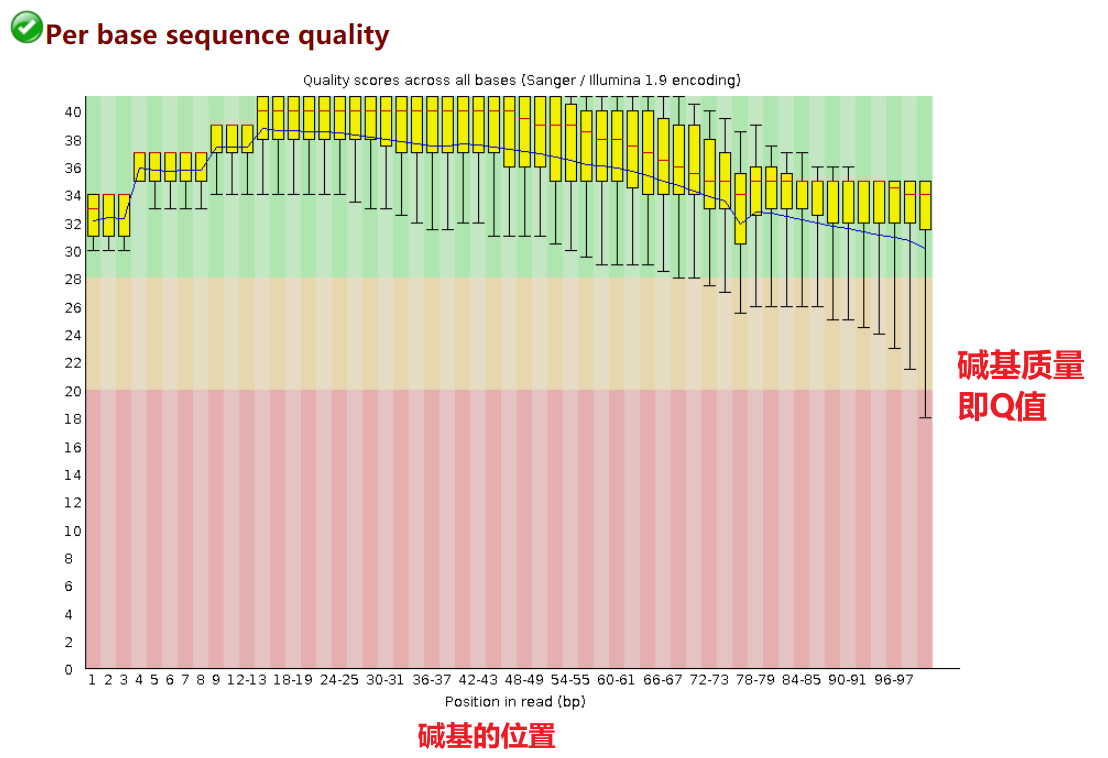
此图代表各个位置的碱基质量均在可接受范围内:Q30以上。
低质量数据如下所示: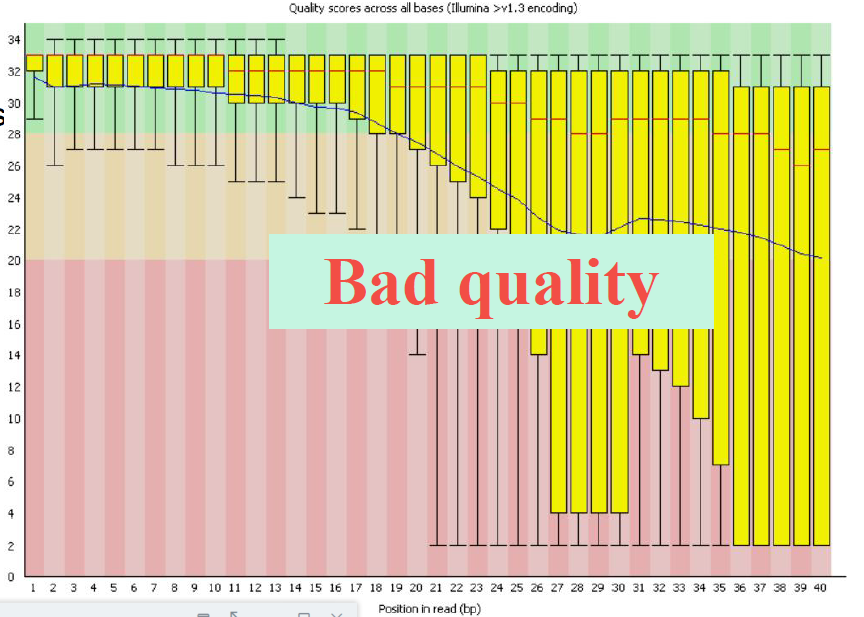
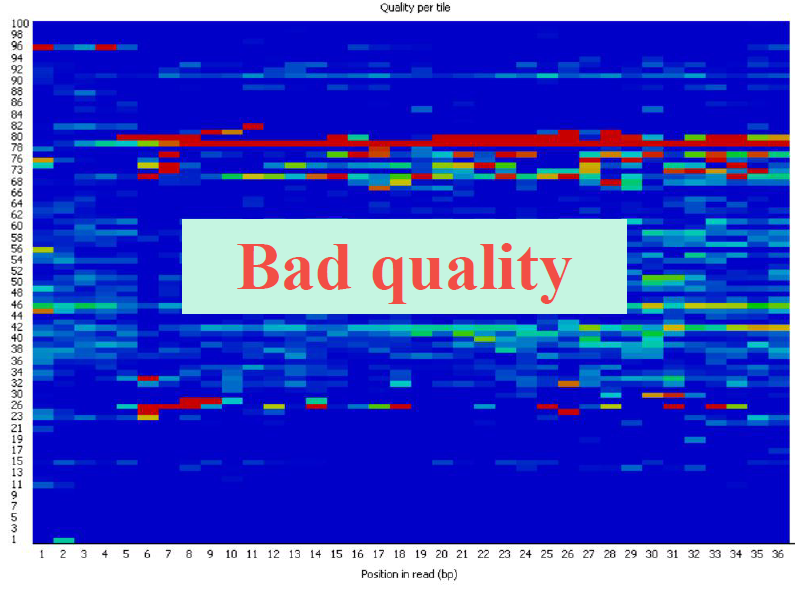
Per Tile Sequence Quality

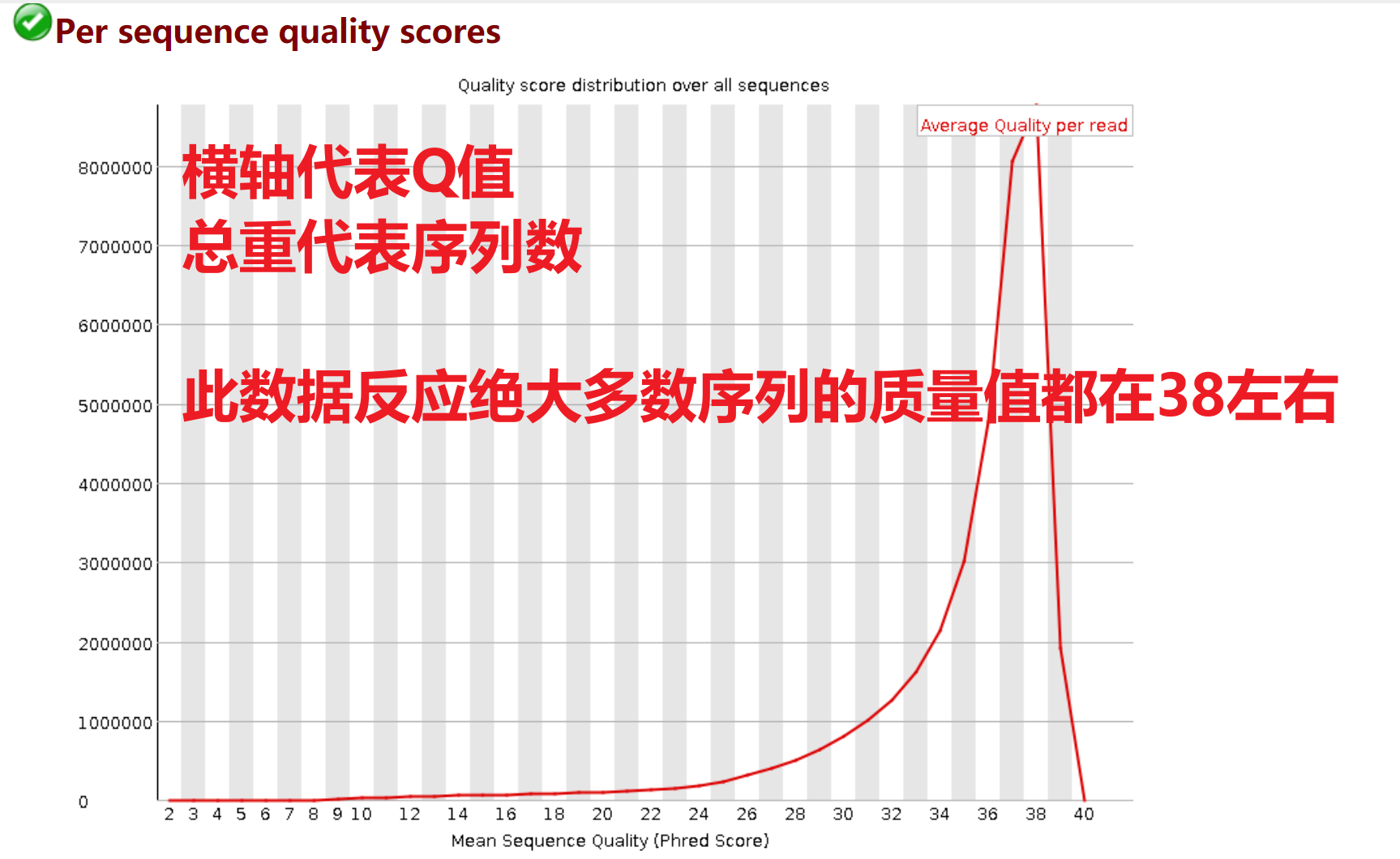
Per Sequence Quality Scores
Per Base Sequence Content
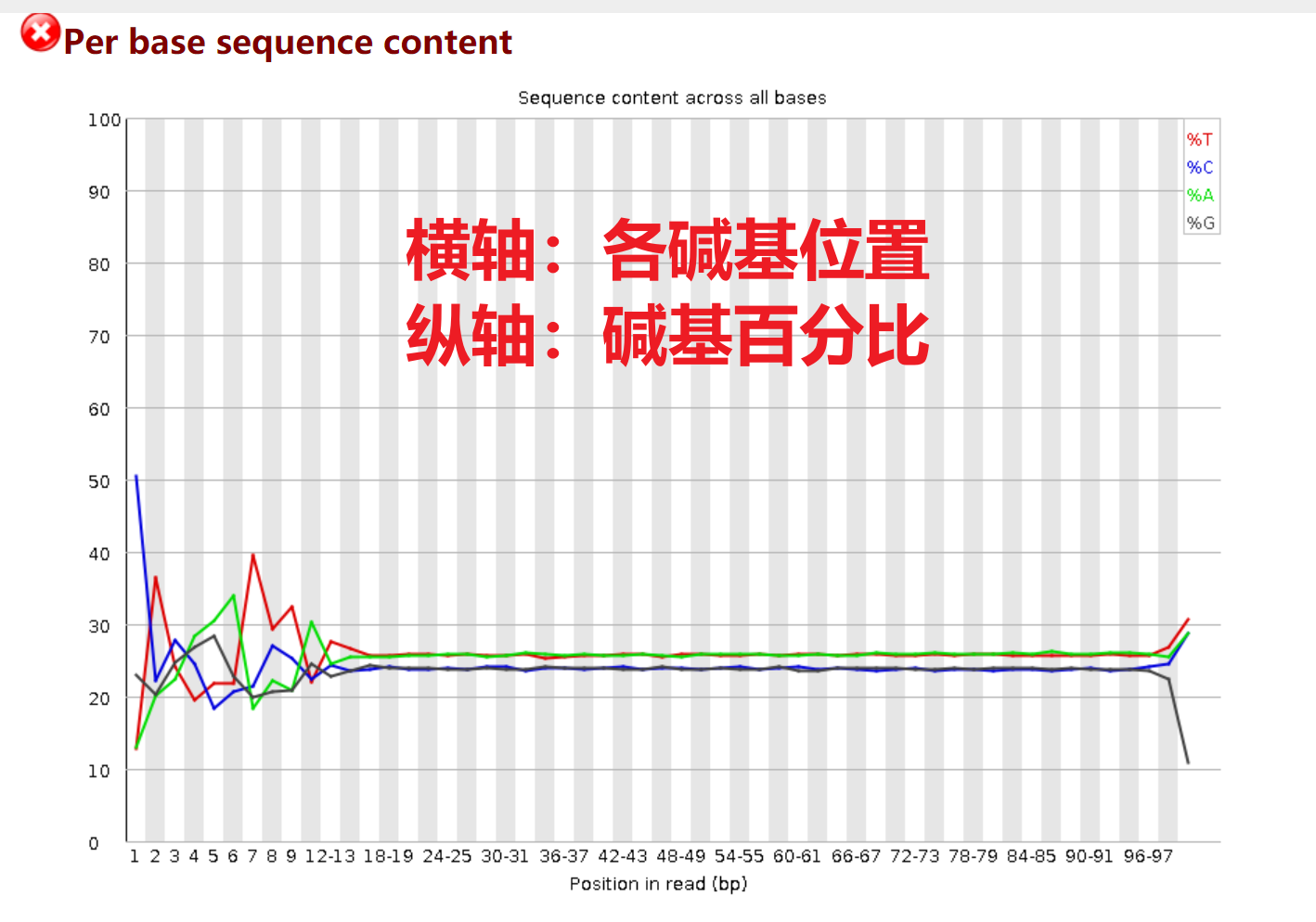
这个数据的Per Base Sequence Content指标不太好
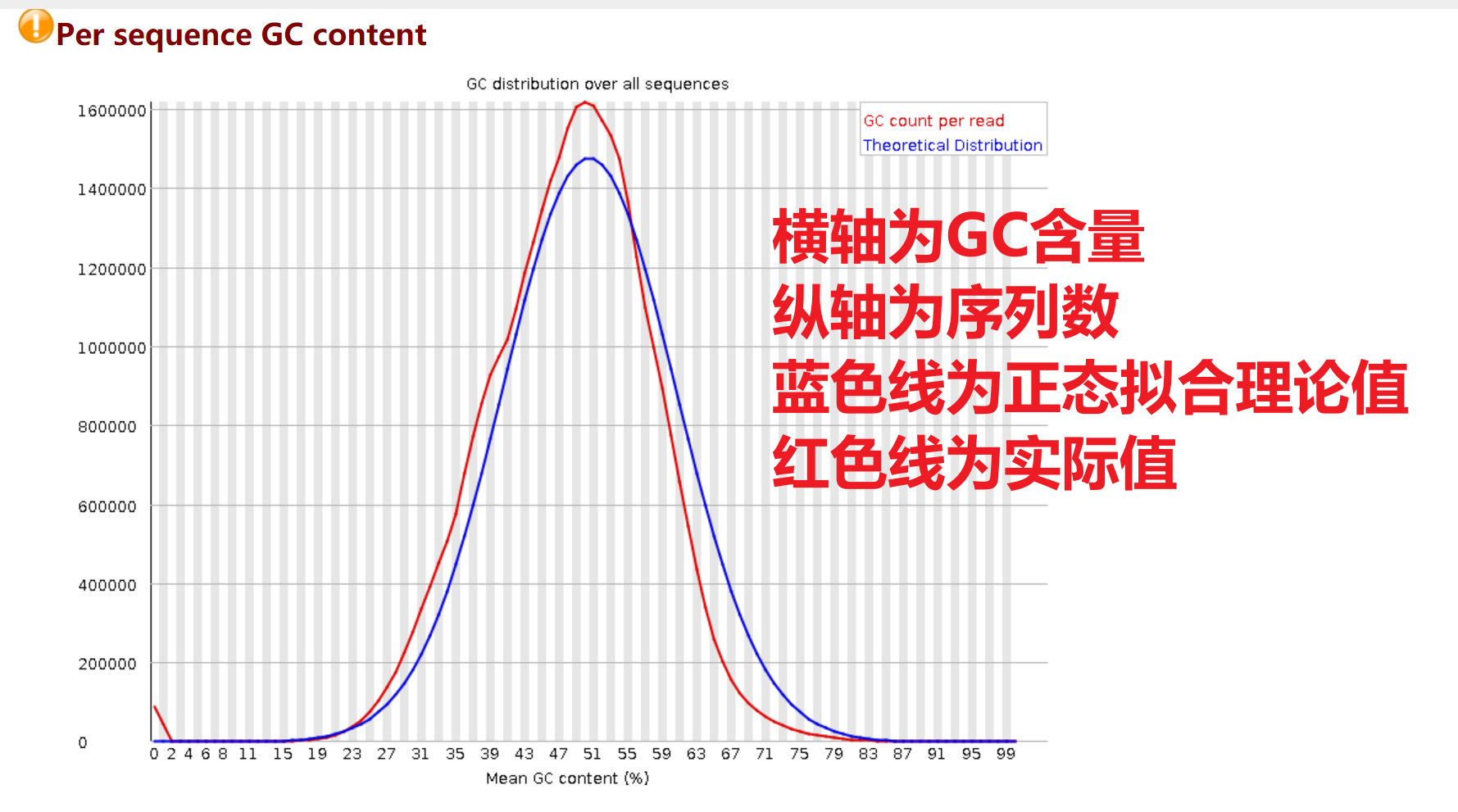
Per sequence GC content
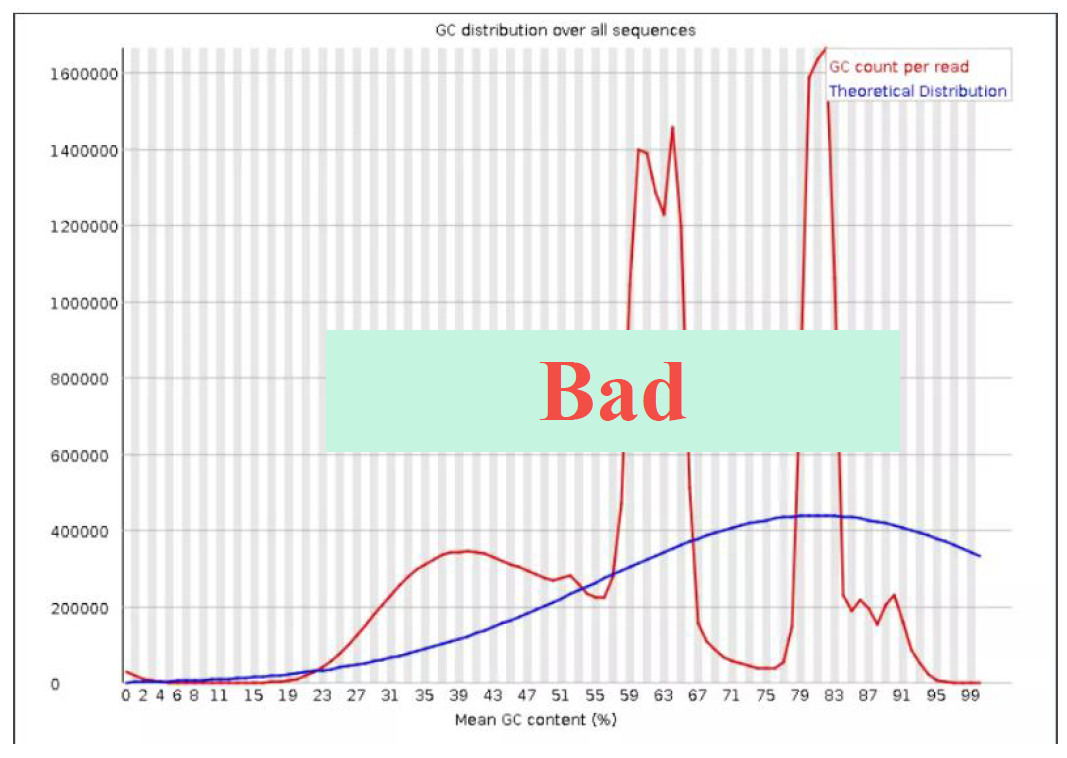
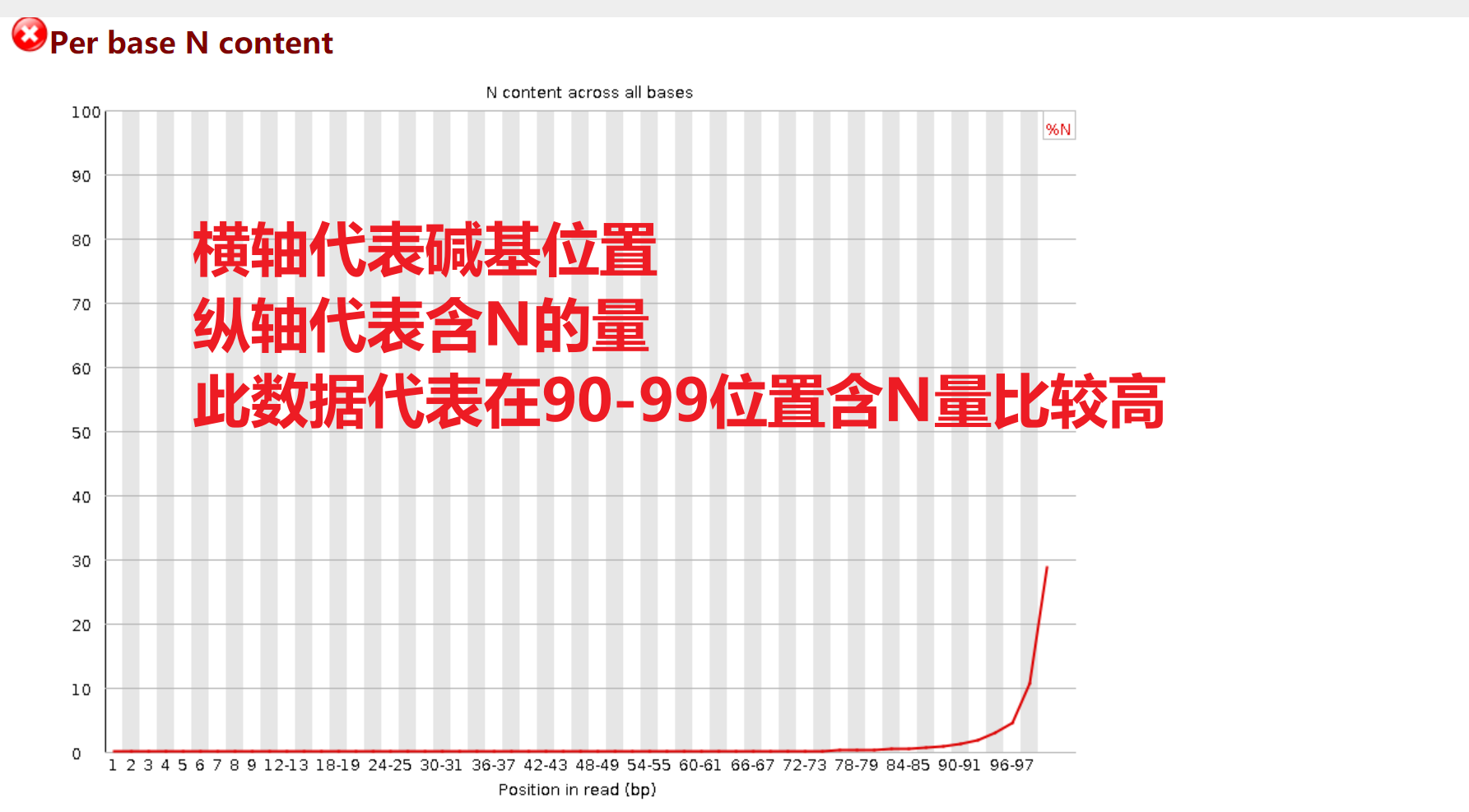
Per base N content
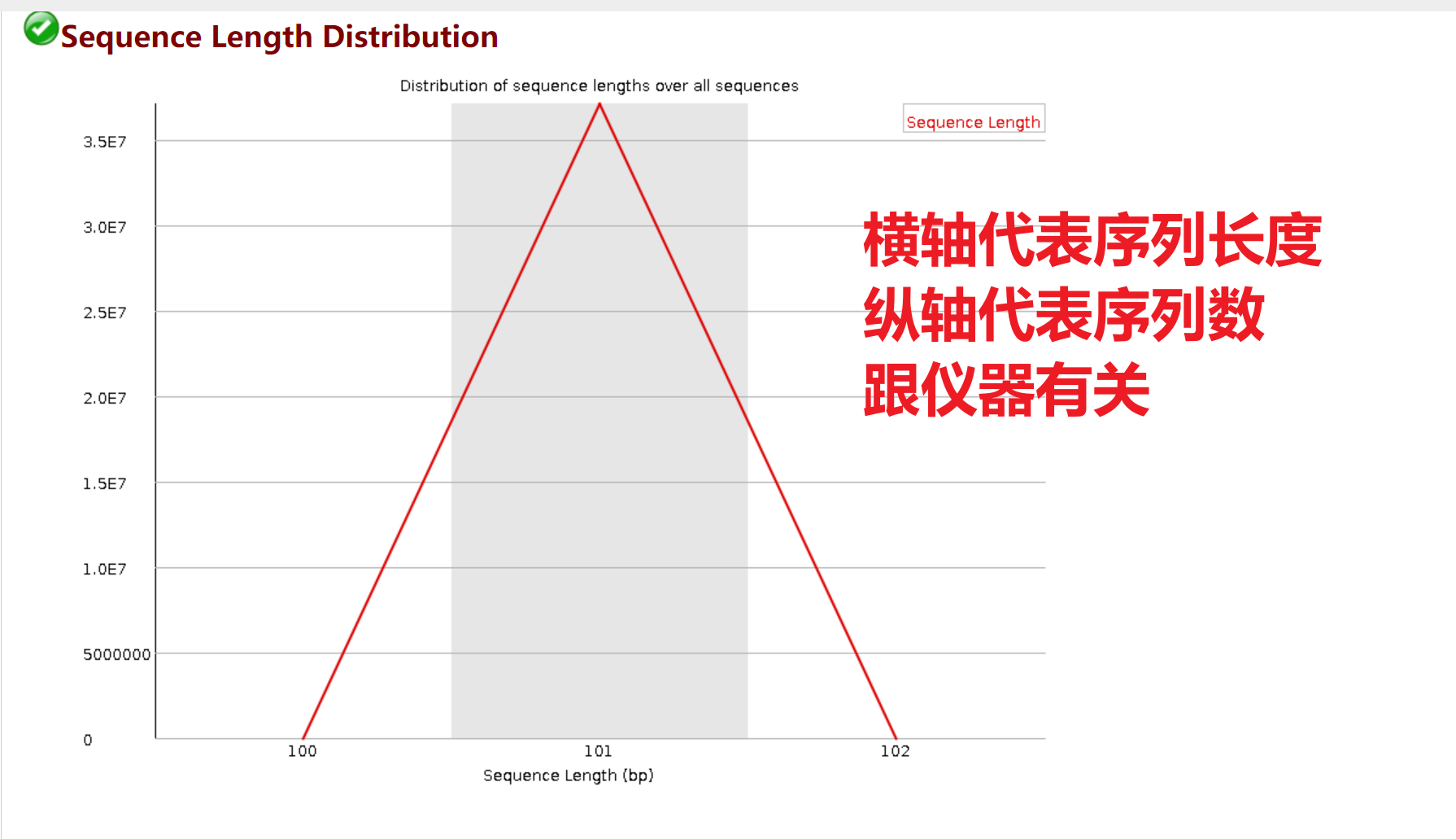
Sequence Length Distribution
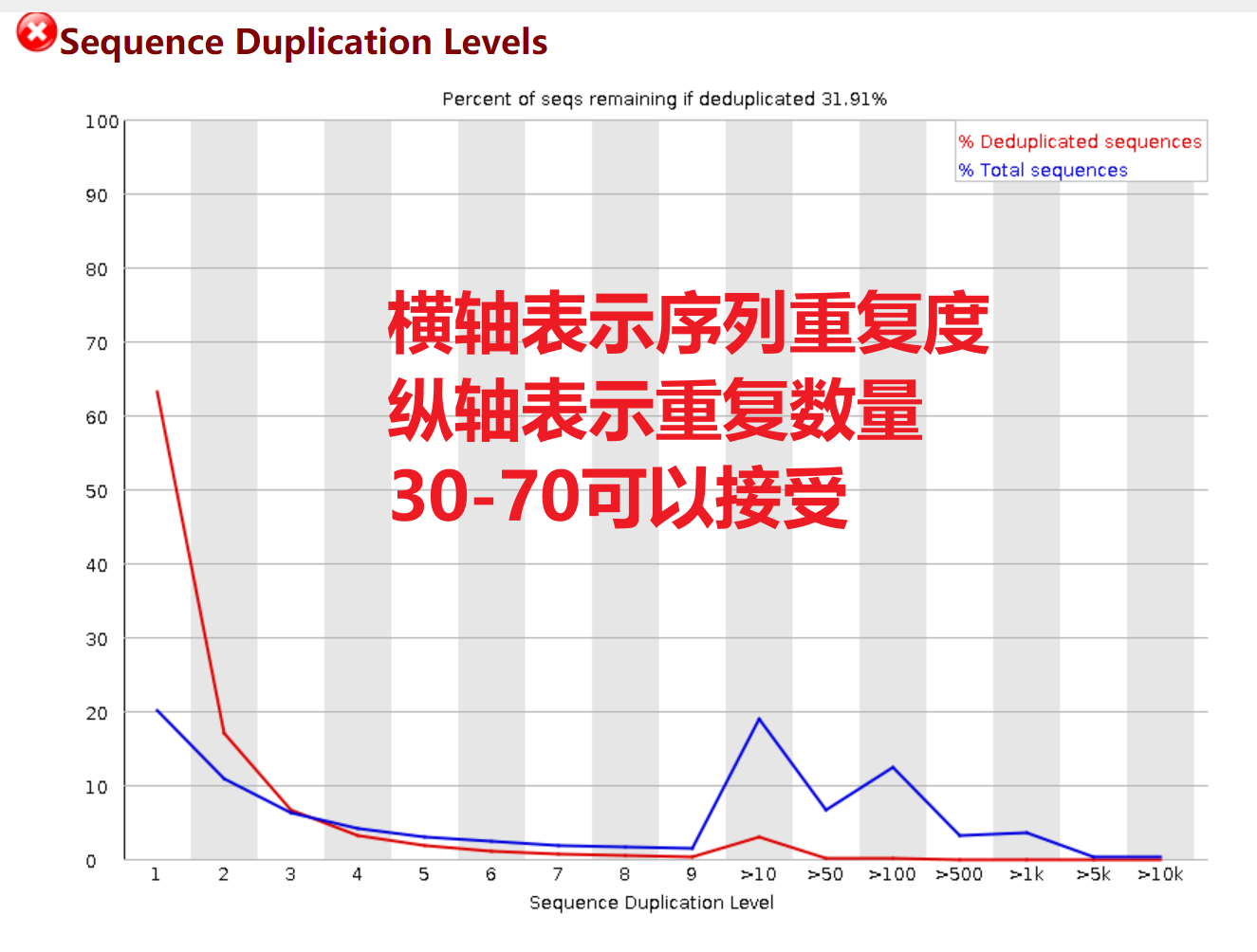
Sequence Duplication Levels
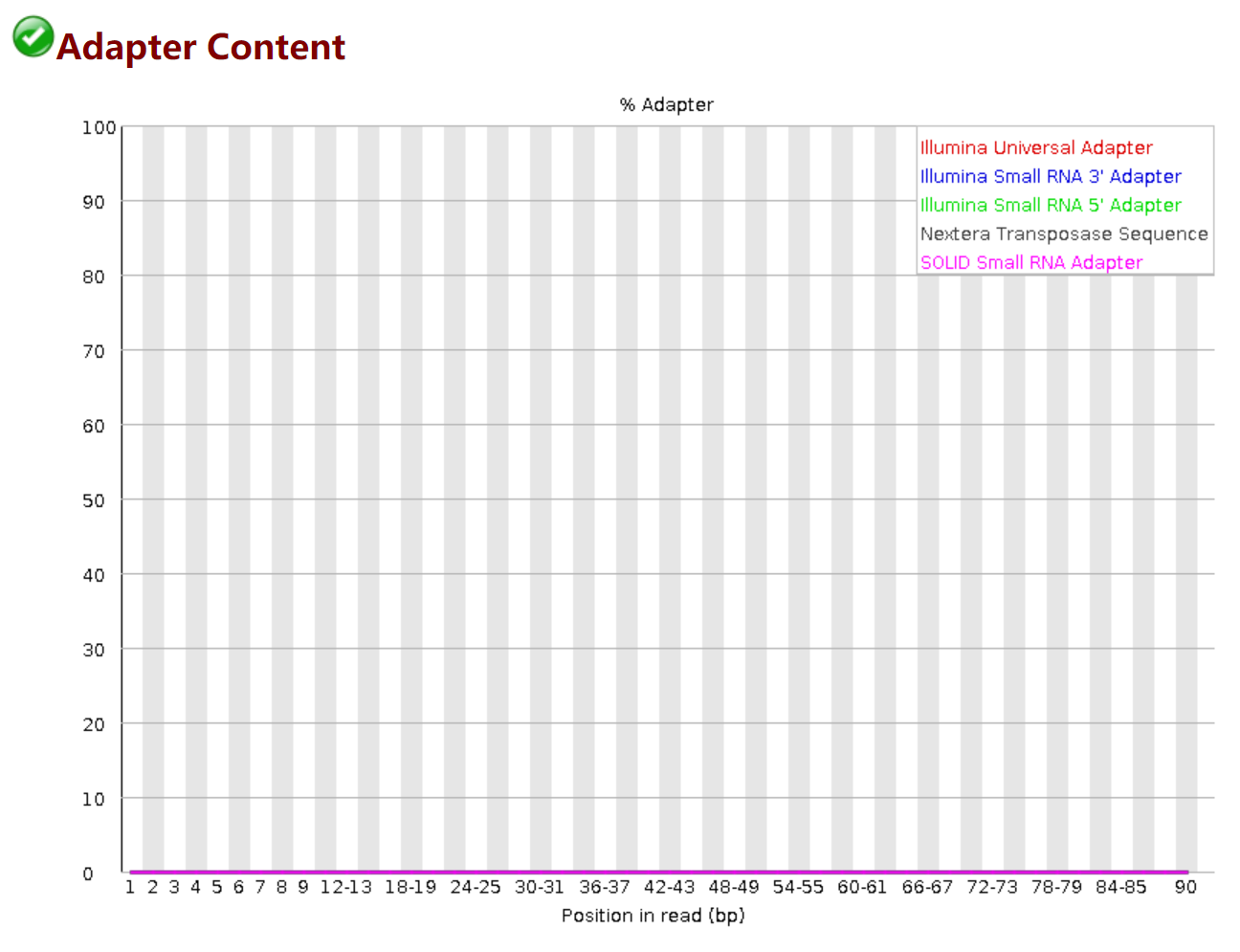
Adapter Content
更多参考:https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
MultiQC报告
为了综合评估所有的样本,使用multiqc命令整合
整合前:
代码
multiqc *zip
整合后:

若有收获,就点个赞吧
0 人点赞
