简介
定义
转录组(transcriptome)广义上指某一生理条件下,细胞内所有转录产物的集合,包括信使RNA、核糖体RNA、转运RNA及非编码RNA;狭义上指所有mRNA的集合。
除转录组外,还有其它一系列的组学: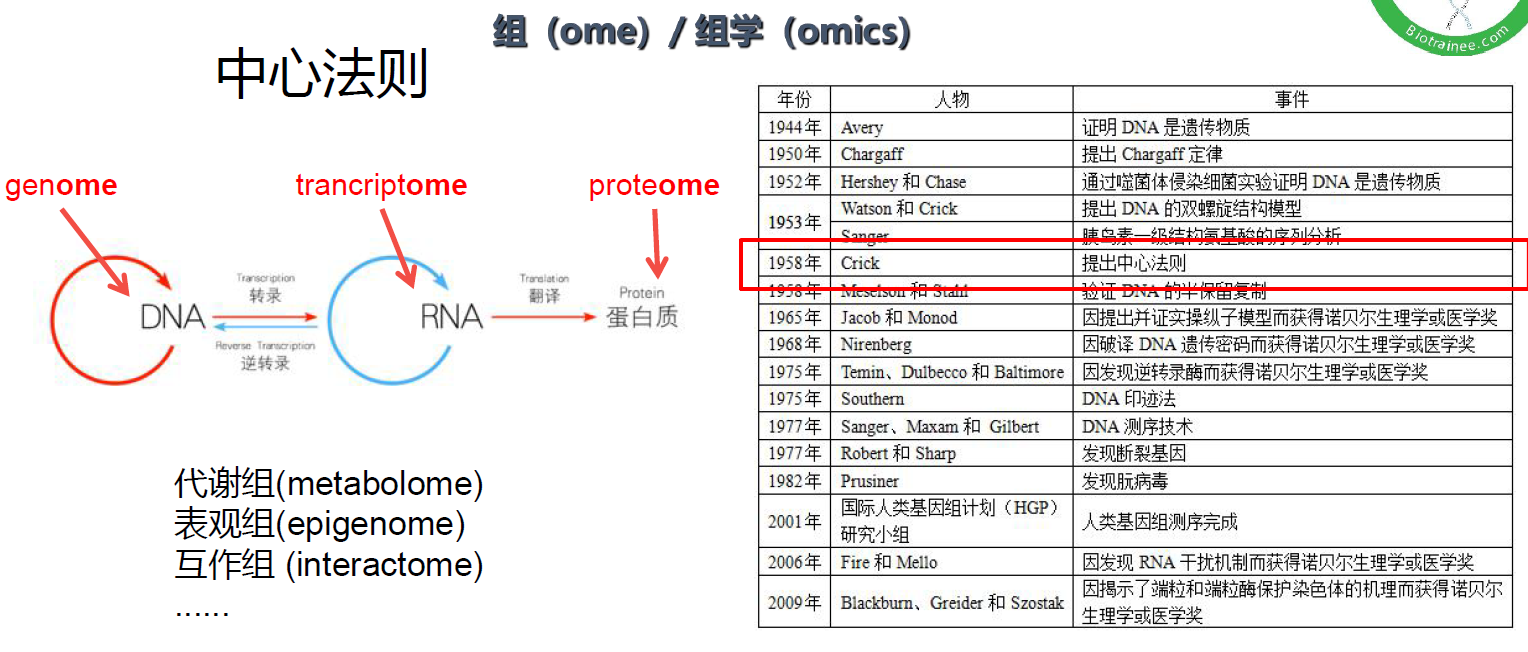
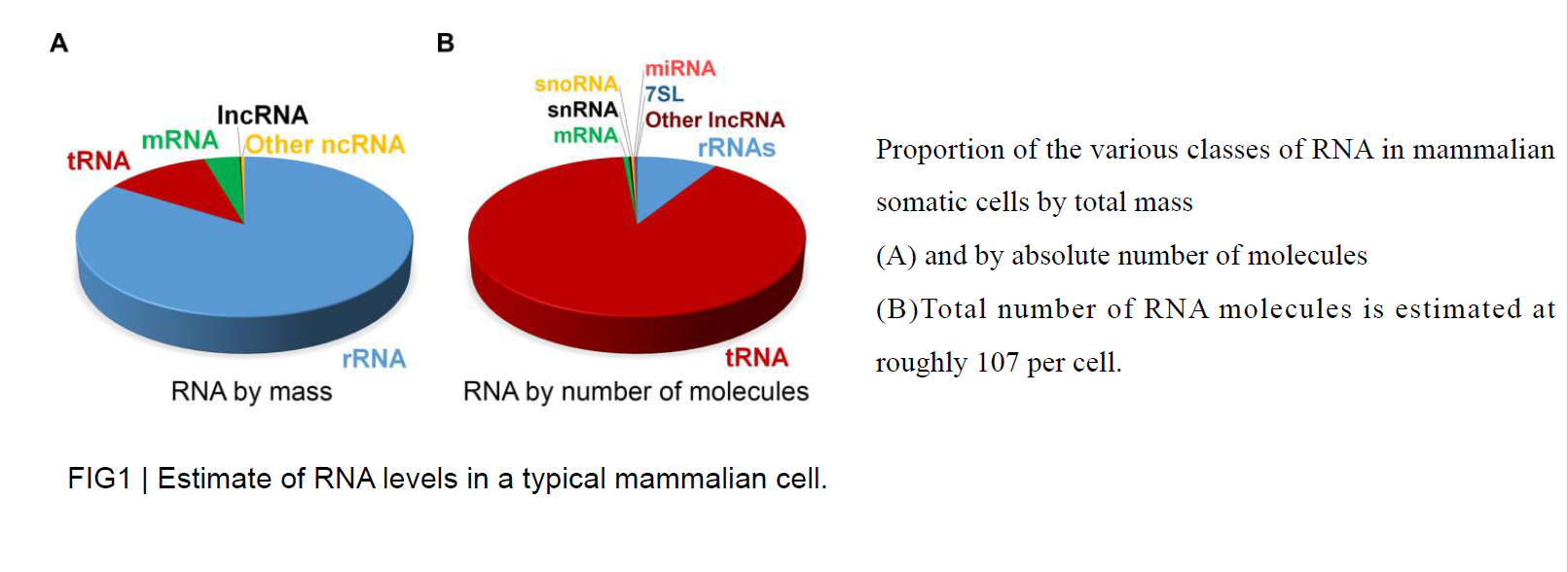
RNA
经典文献
- Conesa A, Madrigal P, Tarazona S, Gomez-Cabrero D, Cervera A, McPherson A, Szcześniak MW, Gaffney DJ, Elo LL, Zhang X, Mortazavi A. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016 Jan 26;17:13. doi: 10.1186/s13059-016-0881-8. Erratum in: Genome Biol. 2016;17(1):181. PMID: 26813401; PMCID: PMC4728800.
- Stark R, Grzelak M, Hadfield J. RNA sequencing: the teenage years. Nat Rev Genet. 2019 Nov;20(11):631-656. doi: 10.1038/s41576-019-0150-2. Epub 2019 Jul 24. PMID: 31341269.
转录组实验流程
样品检测
高质量的RNA是整个项目成功的基础。使用以下方法对样品进行检测,检测结果达到要求后方可进行建库:
- Nanodrop检测RNA的纯度(OD260/280)、浓度、核酸吸收峰是否正常;
- Agilent 2100精确检测RNA的完整性,检测指标包括:RIN值、28S/18S、图谱基线有无上抬、5S峰。
对于降解样本难以获取完整的转录本信息,影响数据质量及完整性。
当RNA总量较低时,会导致建库成功率低,或数据dup率高等问题。
文库构建
样品检测合格后,进行文库构建,主要流程如下:
- 磁珠富集真核生物mRNA(此步骤对RNA的完整性要求比较高,一般RIN值要大于8);
- mRNA进行随机打断;
- 以mRNA为模板,合成第一条cDNA链和第二条cDNA链
- 进行末端修复、加A尾并连接测序接头,然后进行片段大小选择;
- 最后通过PCR富集得到cDNA文库。
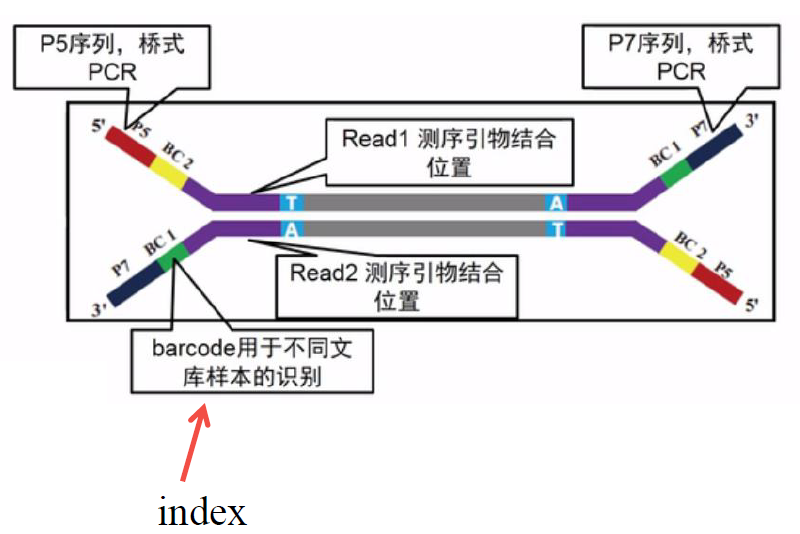
文库:**连接好接头的cDNA,叫做文库,英文为library。
Y字接头:自身不配对,可以有效避免接头在连接的过程中自连接,用途是与flowcell上的接头进行连接。
插入的cDNA序列是各种各样的。
index:一段特定的序列,标记不同来源的样本6-8个碱基。
read2测序引物结合位点:**在Index序列的旁边GAT。
文库质控
文库构建完成后,对文库质量进行检测,检测结果达到要求后方可进行上机测序,检测方法如下:
- 使用Qubit2.0进行初步定量,使用Agilent 2100对文库的插入片段(insert size)进行检测,insert size符合预期后才可进行下一步实验。
- Q-PCR方法对文库的有效浓度进行准确定量(文库有效浓度>2nM),完成库检。
上机测序
二代测序原理:边合成边测序。https://www.bilibili.com/video/BV1ht411q7Wh?from=search&seid=7941223426217329780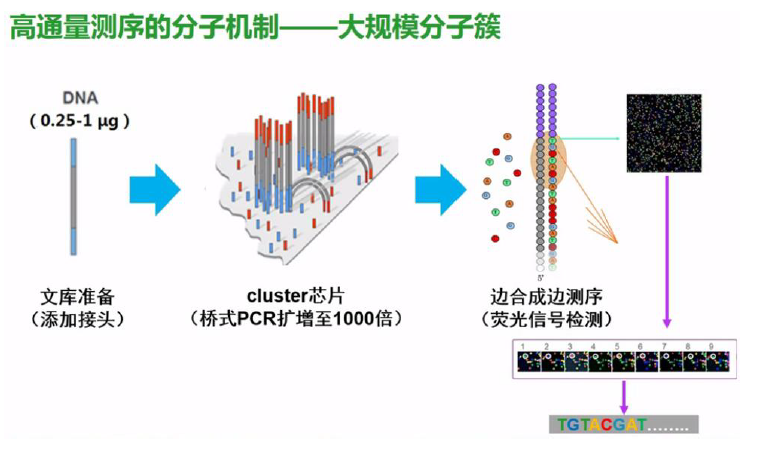
二代测序原理
SBS(Sequencing-By-Synthesis)
通过单分子阵列实现在小型芯片(Flowcell)上进行桥式PCR反应。通过可逆阻断技术实现每次只合成一个碱基,再利用四种带有不同荧光标记的碱基,通过荧光激发/捕获,读取碱基信息。基于可逆终止的、荧光标记dNTP,边合成边测序。流动池
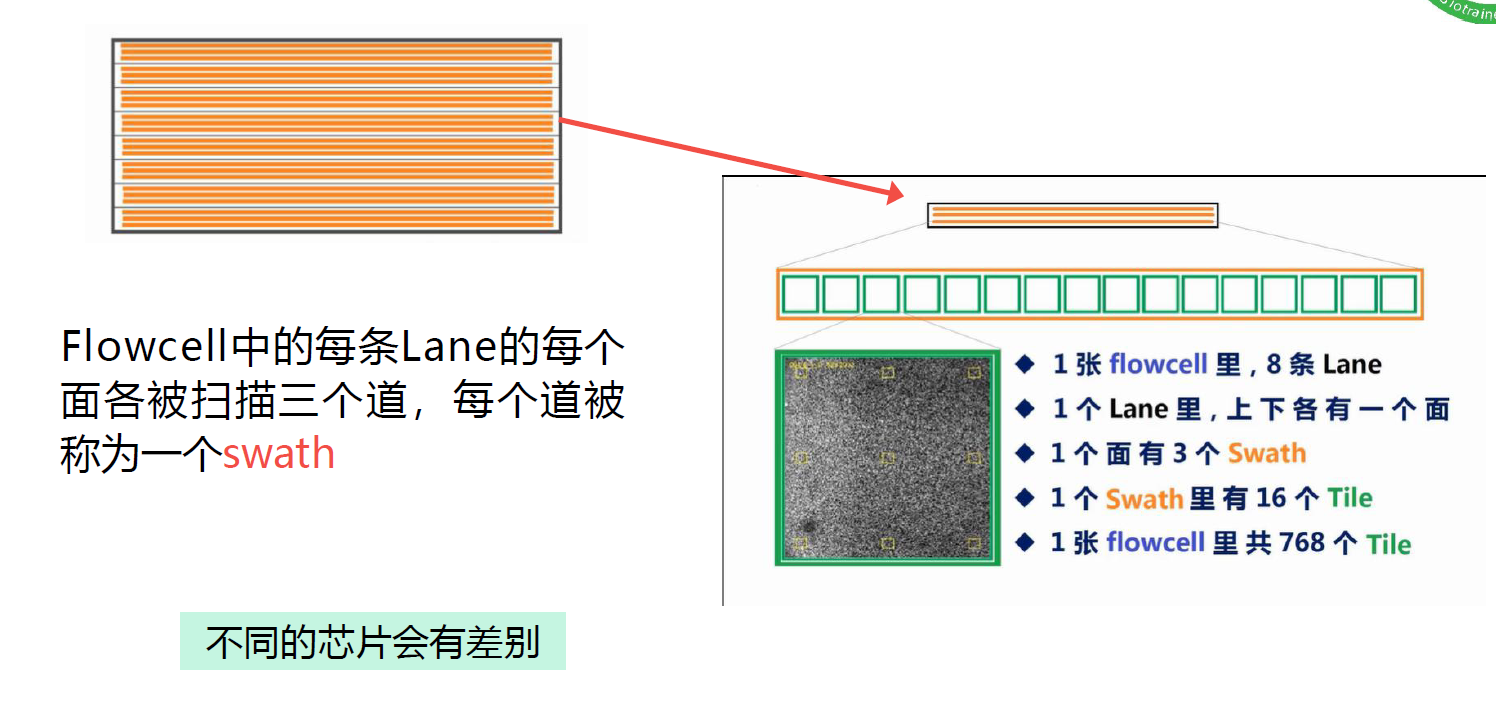
- 芯片(8条通道-lane):内表面做了专门的化学修饰,布满了短的oligo序列(P7/P5接头)2中DNA引物,种在玻璃表面,通过共价键连接。
- 液流孔:每个lane的两端,液流流进、流出的地方。
- swath:Flowcell中的每条Lane的每个面各被扫描三个道,每个道被称为一个swath

桥式PCR
把文库种到芯片上去,然后扩增,文库两头的DNA序列与芯片上的引物互补,互补杂交杂交完后,加入dNTP和聚合酶,合成双链,加入NaOH碱溶液,双链解开,加入中性液体,环境变成中性。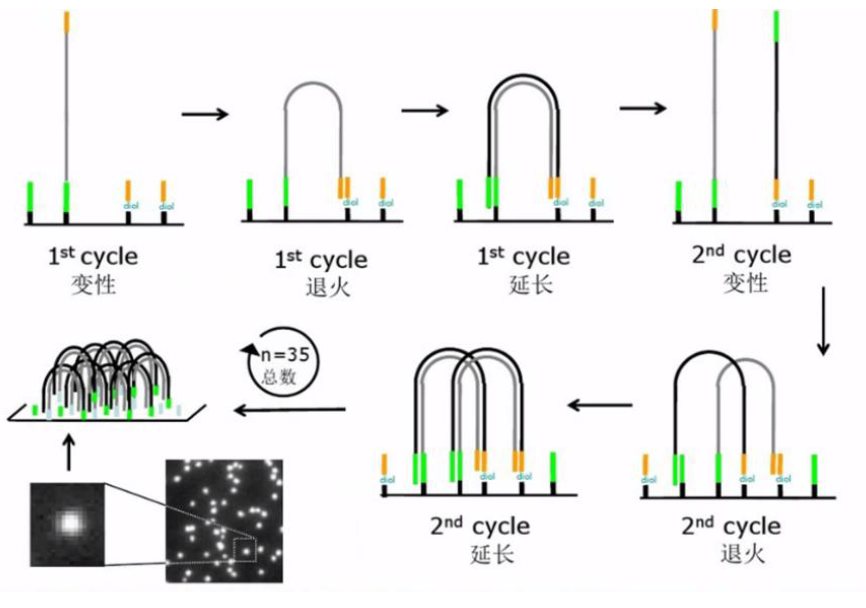
转录组分析策略
标准分析流程
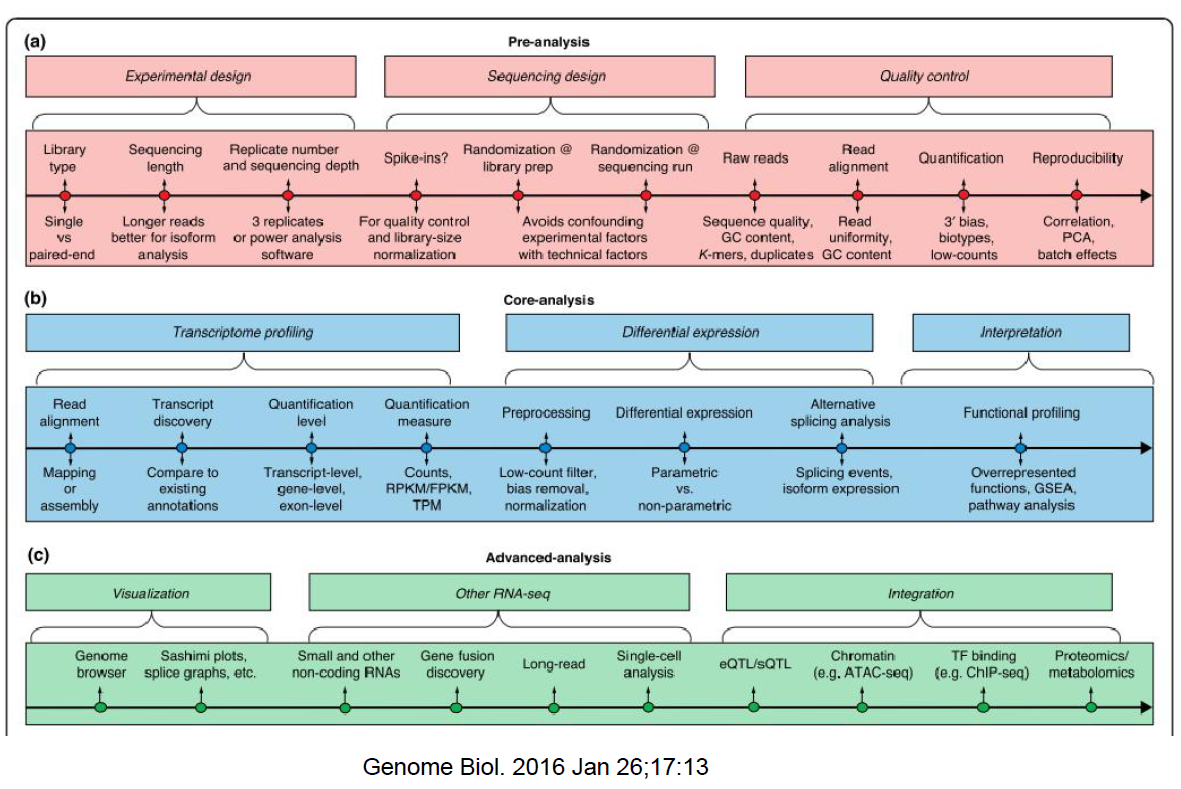
分析前处理:实验设计、分析内容设计、质量控制等。
核心分析结果:转录组图谱、差异表达分析、功能富集分析。
进阶分析:可视化、miRNA、m6A分析、单细胞分析、互作组学等。
RNA-seq流程
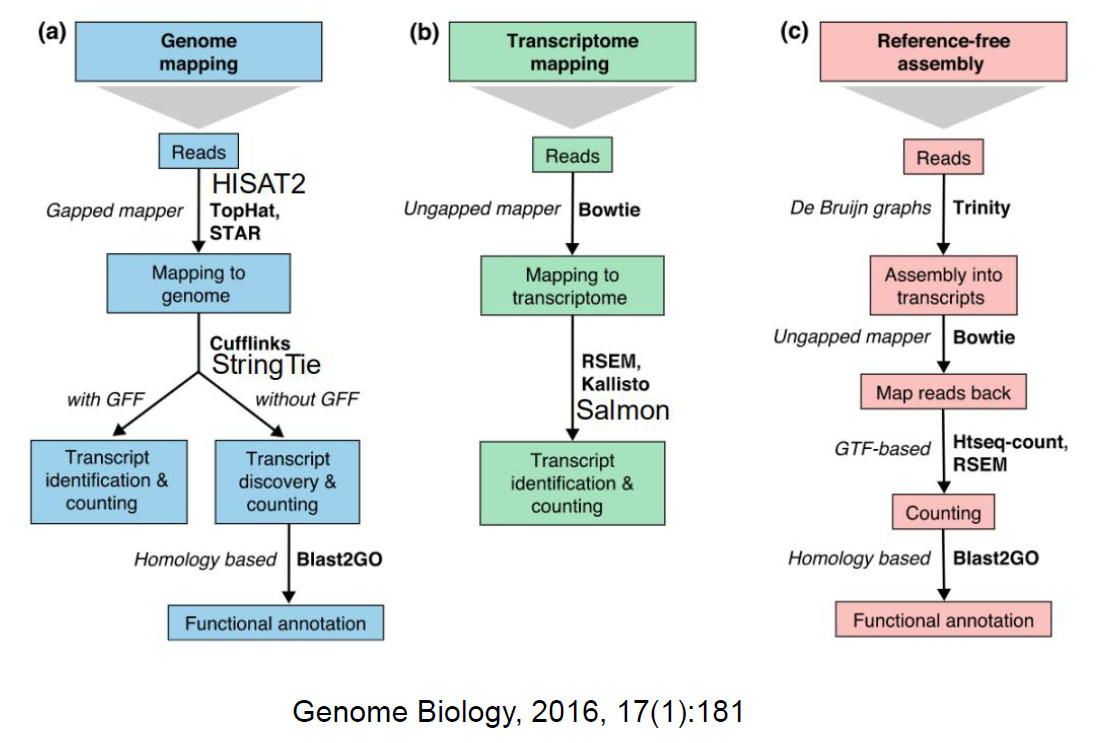
有参考序列ab的RNA-seq:得到reads—做比对—定量—功能分析(差异、富集)。
未知参考序列c:新的物种研究等。
常见数据格式
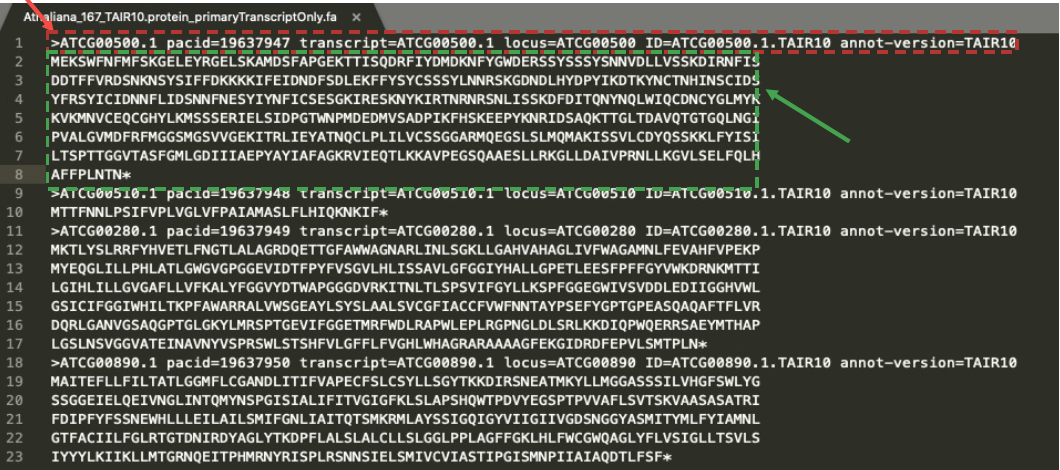
fasta
是一种基于文本用于表示核酸序列或多肽序列的格式。其中核酸或氨基酸均以单个字母来表示,且允许在序列前添加序列名及注释。
特征:分为两部分(ID行和序列行)
- ID行:以>开头,有时会包含注释信息。
- 序列行:表示一个碱基或者氨基酸,直到下一个ID行为止。
fastq
是一种存储了生物序列以及相应的质量评价的文本格式。
特征:4行(ID行、序列行、附加信息行、碱基质量行)
- ID行:以@开头,包含必要信息。
- 序列行:包含测序数据。
- 附加信息行:与ID行信息一致,或者为+号。
- 碱基质量行:根据ASCII表,用一个字符表示碱基质量的好坏。
更多请参考:https://support.illumina.com/bulletins/2016/04/fastq-files-explained.html
gff(General Feature Format)
记录序列中转录起始位点、基因、外显子、内含子等组成元件在染色体中的位置信息。
现在用得比较多的是第3版,即gff3。
特征:9列。
- Column 1: seqid。 序列的编号,编号的有效字符[a-zA-Z0-9.:^*$@!=_?-|]
- Column 2: source。注释信息的来源,比如”Genescan”、”Genbank”等,可以为空,为空用”.”点号代替
- Column 3: type。注释信息的类型,比如Gene、cDNA、mRNA等,或者是SO对应的编号
- Columns 4 & 5: start and end。开始与结束的位置,注意计数是从1开始的。结束位置不能大于序列的长度
- Column 6: score。得分,数字,是注释信息可能性的说明,可以是序列相似性比对时的E-values值或者基因预测是的P-values值。”.”表示为空。
- Column 7: strand。序列的方向, +表示正义链, -反义链 , ? 表示未知.
- Column 8: phase。仅对注释类型为 “CDS”有效,表示起始编码的位置,有效值为0、1、2。
- Column 9: attributes。以多个键值对组成的注释信息描述,键与值之间用”=”,不同的键值用”;”隔开,一个键可以有多个值,不同值用”,”分割。
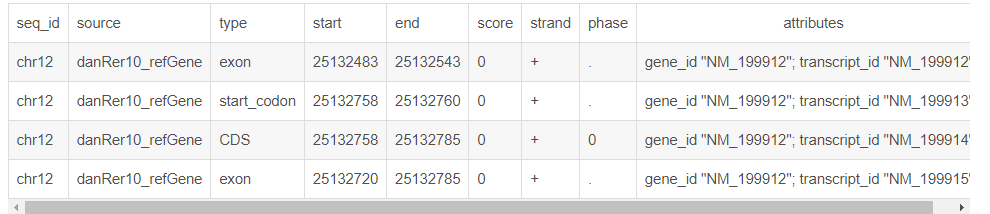
gtf(Gene Transfer Format)
GTF全称为 gene transfer format ,主要是用来对基因进行注释。
特征:9列。
- 1) seq_id:序列的编号,一般为chr或者scanfold编号;
- 2) source: 注释的来源,一般为数据库或者注释的机构,如果未知,则用点“.”代替;
- 3) type: 注释信息的类型,比如Gene、cDNA、mRNA、CDS等
- 4) start: 该基因或转录本在参考序列上的起始位置;
- 5) end: 该基因或转录本在参考序列上的终止位置;
- 6) score: 得分,数字,是注释信息可能性的说明,可以是序列相似性比对时的E-values值或者基因预测是的P-values值,“.”表示为空;
- 7) strand: 该基因或转录本位于参考序列的正链(+)或负链(-)上;
- 8) phase: 仅对注释类型为“CDS”有效,表示起始编码的位置,有效值为0、1、2(对于编码蛋白质的CDS来说,本列指定下一个密码子开始的位置。每3个核苷酸翻译一个氨基酸,从0开始,CDS的起始位置,除以3,余数就是这个值,,表示到达下一个密码子需要跳过的碱基个数。该编码区第一个密码子的位置,取值0,1,2。0表示该编码框的第一个密码子第一个碱基位于其5’末端;1表示该编码框的第一个密码子的第一个碱基位于该编码区外;2表示该编码框的第一个密码子的第一、二个碱基位于该编码区外;如果Feature为CDS时,必须指明具体值。);
- 9) attributes: 一个包含众多属性的列表,格式为“标签=值”(tag=value),标签与值之间以空格分开,且每个特征之后都要有分号;(包括最后一个特征),其内容必须包括gene_id和transcript_id。以多个键值对组成的注释信息描述,键与值之间用“=”,不同的键值用“;

若有收获,就点个赞吧
0 人点赞
