基因组数据库
常用的参考基因组数据库有以下三个,按个人习惯选择使用(我喜欢用ENSEMBL)。
http://www.ensembl.org/index.html?redirect=no

https://www.ncbi.nlm.nih.gov/projects/genome/guide/human/index.shtml
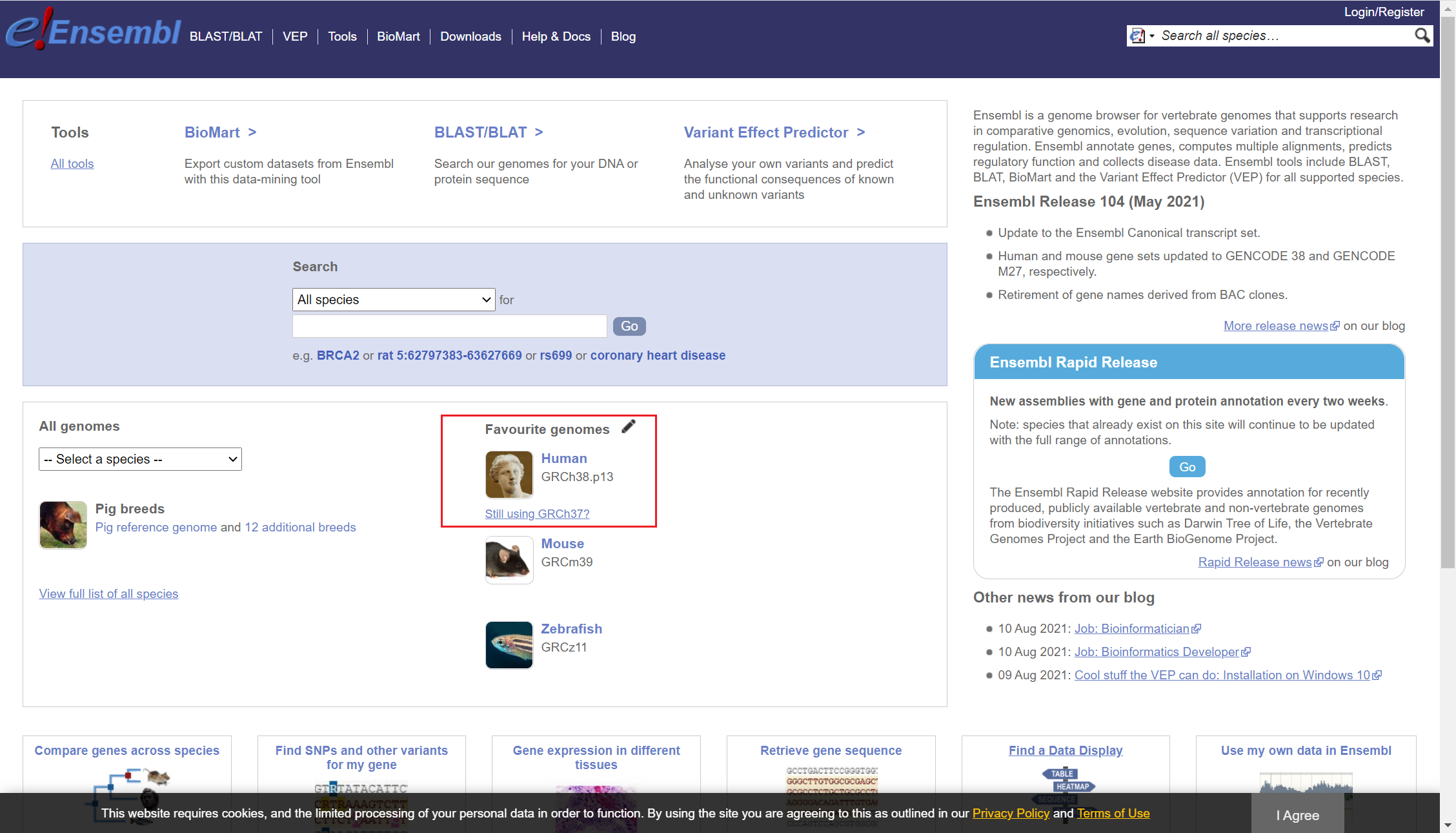
Ensembl
参考基因组下载

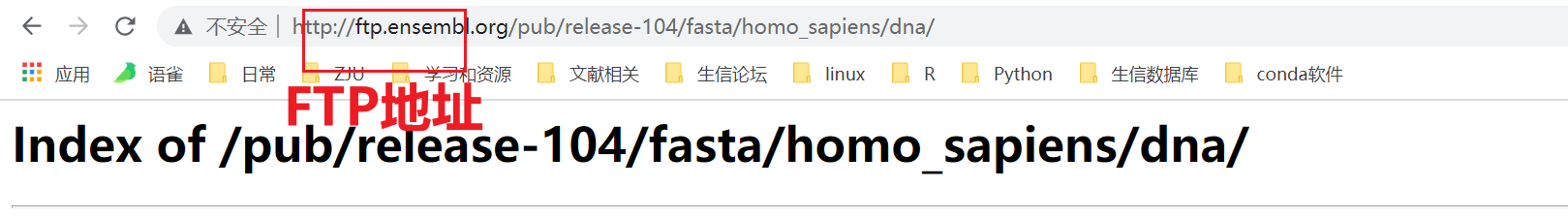
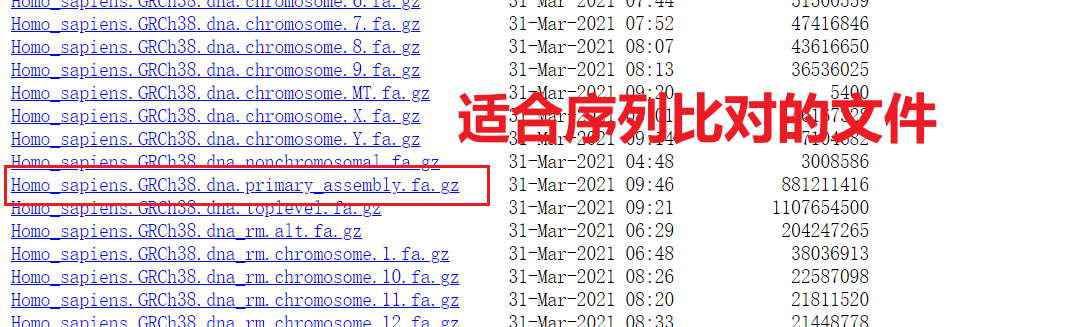
nohup wget -c http://ftp.ensembl.org/pub/release-104/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz &
基因组注释文件下载
注:教程演示,下载人类基因组对比序列和鼠的基因组注释文件(我就缺这两个)。不要照抄代码!真正做分析需要二者匹配。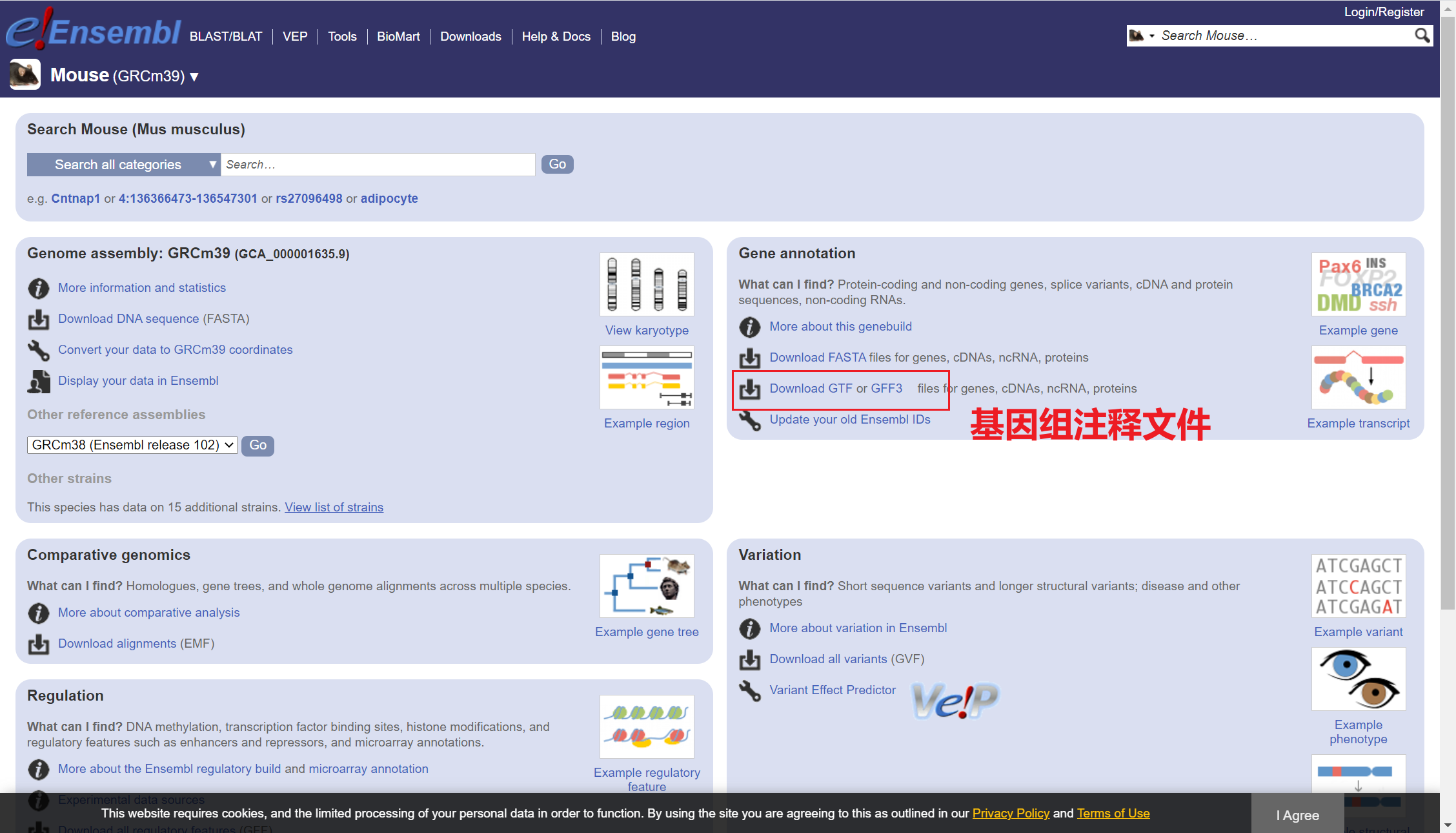
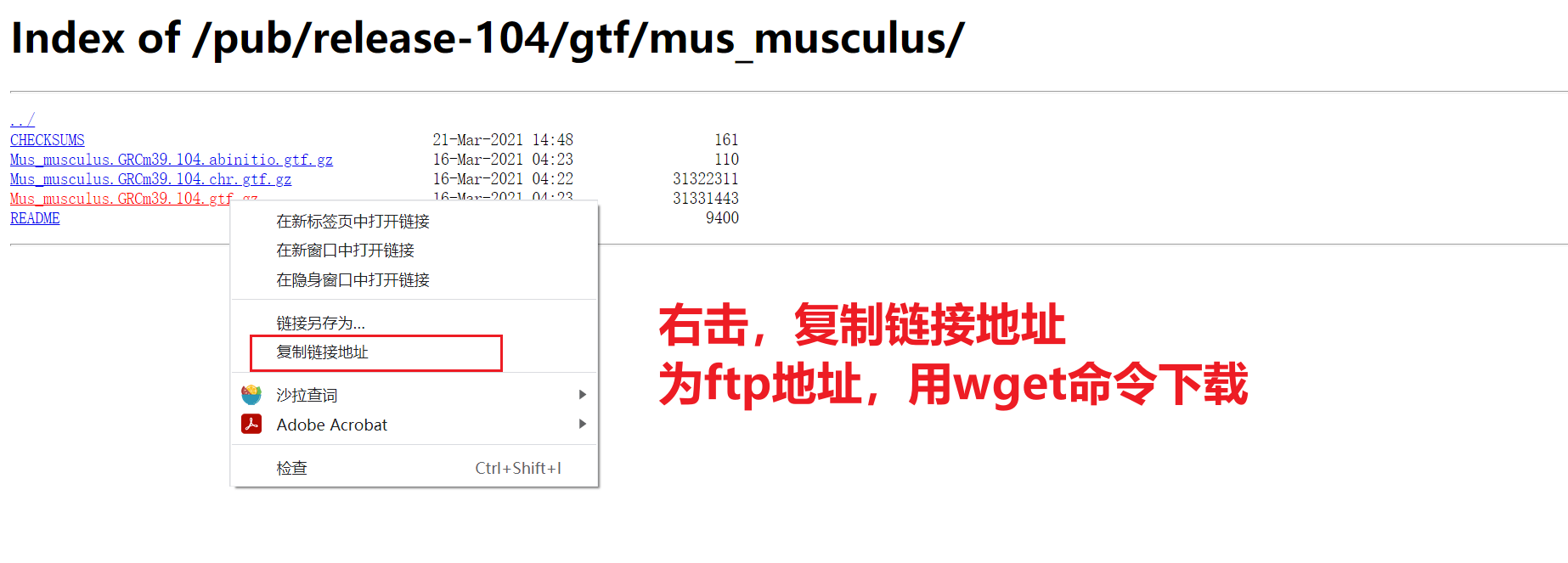
nohup wget -c http://ftp.ensembl.org/pub/release-104/gtf/mus_musculus/Mus_musculus.GRCm39.104.gtf.gz &
cDNA参考序列下载
转录组需要用到cDNA。

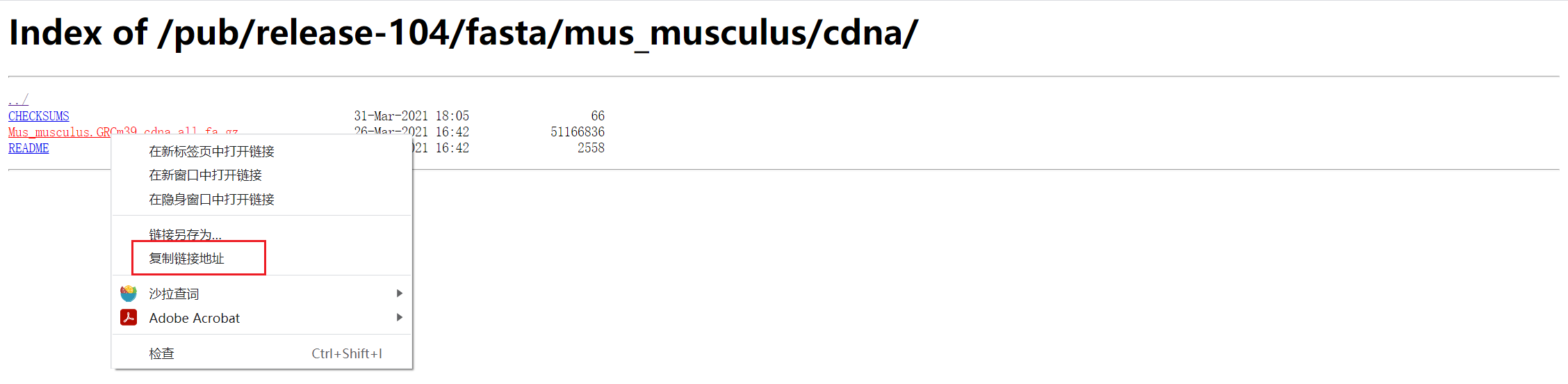
nohup wget -c http://ftp.ensembl.org/pub/release-104/fasta/mus_musculus/cdna/Mus_musculus.GRCm39.cdna.all.fa.gz &
fasta数据格式
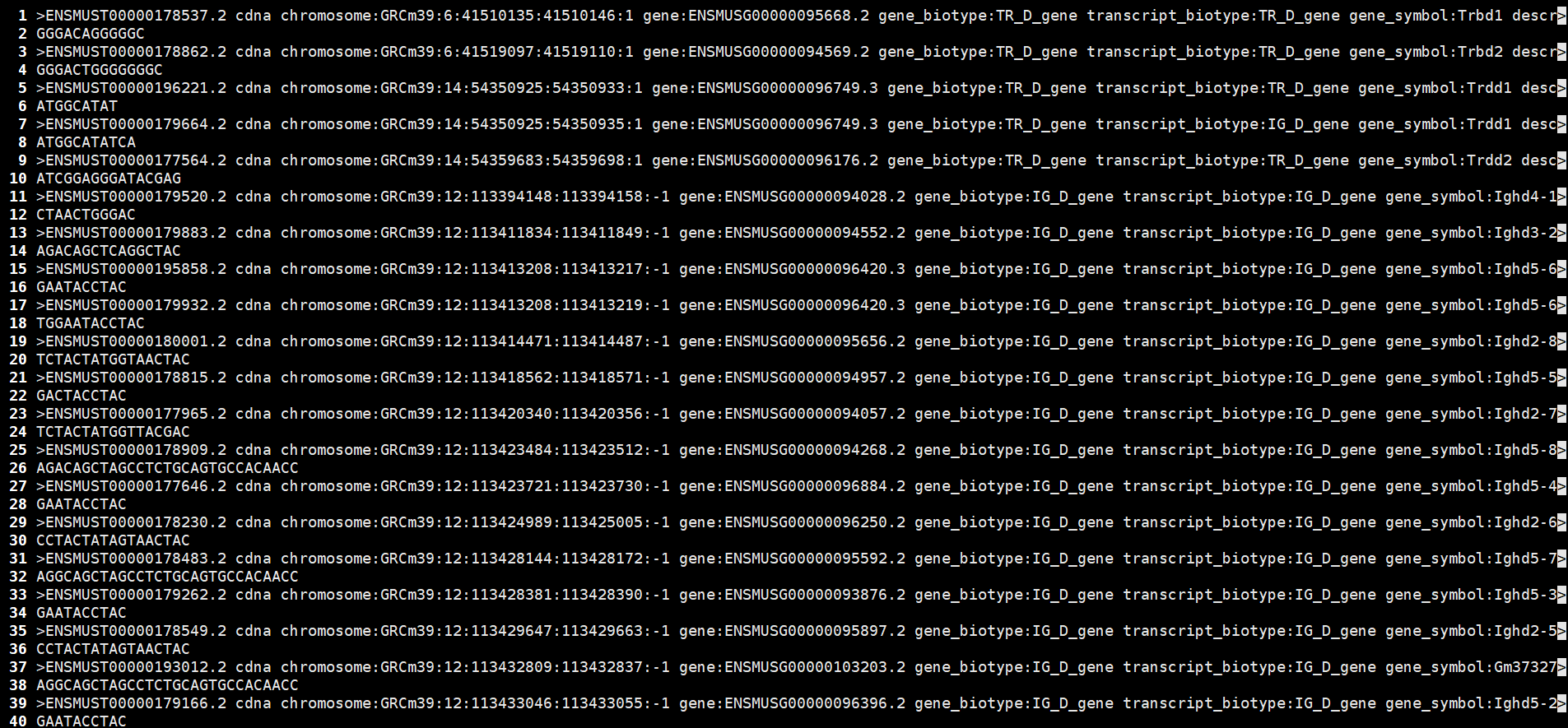
- 一种用于记录核酸序列或肽序列的文本格式,其中的核酸或氨基酸均以单个字母编码呈现。
- 以“>”开头,序列名称&序列描述
序列中允许空格,换行,空行,直到下一个“>”,表示该列结束。
fastq转换为fasta
awk '{if(NR%4 == 1){print ">" substr($0, 2)}} {if(NR%4 == 2){print}}' fastq > fasta
gff文件
格式

共9列。是一种简单的、方便的对于DNA、RNA以及蛋白质序列的特征进行描述的一种数据格式。
- 指示序列从那里到那里是基因,已经成为序列注释的通用格式。
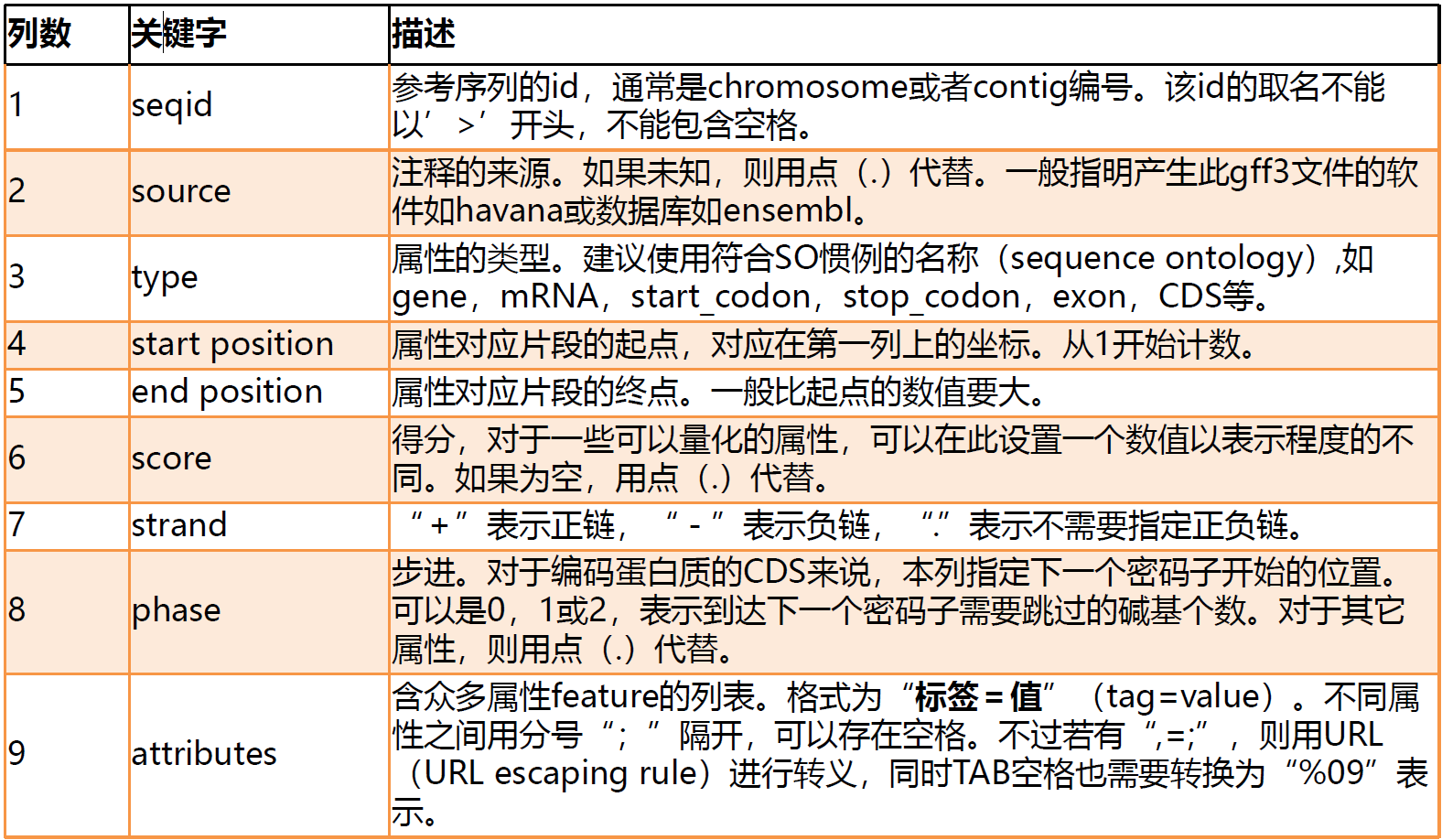
第九列详细介绍
- ID:属性feature的唯一标识,一个GFF文件内ID具有唯一性
- Name:属性feature所展示的名称
- Alias:属性feature的第二个name,可以不具有唯一性
- Parent:属性feature的上一级ID,可以将exons聚集成transcripts,transcripts聚集成genes,and so forth,一个feature可以有多个parents.
- Target:表示比对的目标区域,格式为”target id start end[strand”,其中strand是可选(”+”or”-“)。如果target_id含有空格,必须转换为’%20’
- Gap:比对结果的Gap信息
- Note:文本描述
- Ontology term A reference to an ontology term:对应的GO数据库ID
-
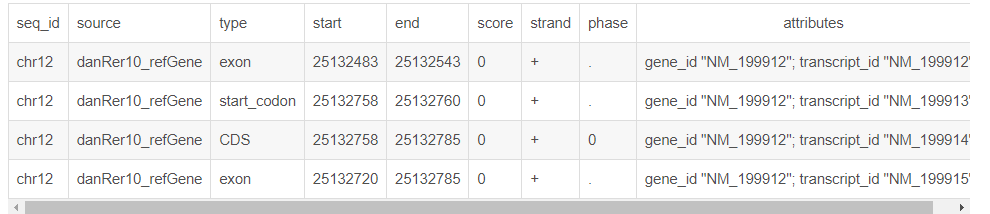
gtf文件
GTF全称为 gene transfer format ,主要是用来对基因进行注释。
特征:9列。 1) seq_id:序列的编号,一般为chr或者scanfold编号;
- 2) source: 注释的来源,一般为数据库或者注释的机构,如果未知,则用点“.”代替;
- 3) type: 注释信息的类型,比如Gene、cDNA、mRNA、CDS等
- 4) start: 该基因或转录本在参考序列上的起始位置;
- 5) end: 该基因或转录本在参考序列上的终止位置;
- 6) score: 得分,数字,是注释信息可能性的说明,可以是序列相似性比对时的E-values值或者基因预测是的P-values值,“.”表示为空;
- 7) strand: 该基因或转录本位于参考序列的正链(+)或负链(-)上;
- 8) phase: 仅对注释类型为“CDS”有效,表示起始编码的位置,有效值为0、1、2(对于编码蛋白质的CDS来说,本列指定下一个密码子开始的位置。每3个核苷酸翻译一个氨基酸,从0开始,CDS的起始位置,除以3,余数就是这个值,,表示到达下一个密码子需要跳过的碱基个数。该编码区第一个密码子的位置,取值0,1,2。0表示该编码框的第一个密码子第一个碱基位于其5’末端;1表示该编码框的第一个密码子的第一个碱基位于该编码区外;2表示该编码框的第一个密码子的第一、二个碱基位于该编码区外;如果Feature为CDS时,必须指明具体值。);
- 9) attributes: 一个包含众多属性的列表,格式为“标签=值”(tag=value),标签与值之间以空格分开,且每个特征之后都要有分号;(包括最后一个特征),其内容必须包括gene_id和transcript_id。以多个键值对组成的注释信息描述,键与值之间用“=”,不同的键值用“;
课后习题

Answer
zless -S Homo_sapiens.GRCh38.95.gtf.gz |awk -F'\t' '{if($3=="gene"){print$9}}' |awk -F';' '{print$1,$3,$5}' |awk '{print$2"\t"$4"\t"$6}' |sed 's/"//g' |grep 'protein_coding' >protein_coding_id2name.xls

若有收获,就点个赞吧
0 人点赞
