撰文:胡海飞特约审稿:杨金良制版编辑:郝樾
泛基因组(Pangenome)是最近比较热门的一个研究方向。Pan,源自希腊语,就是全部的意思。泛基因组,研究的是某一物种全部基因的总和。这次推文就和大家一起认识一下泛基因组,并且快速了解一下该领域的最新进展。
什么是泛基因组?
2005年,Tettelin等人提出了微生物泛基因组概念(pangenome,pan源自希腊语“παν”,全部的意思),泛基因组即某一物种全部基因的总称。2009 年,Li等人首次采用de novo全基因组组装方法对多个人类个体基因组进行拼接,发现了个体独有的DNA序列和功能基因,并首次提出了“人类泛基因组”的概念,即人类群体基因序列的总和。2009 年泛基因组测序被首次应用于人类基因组学研究;2013 年泛基因组测序开始被应用于动植物研究领域。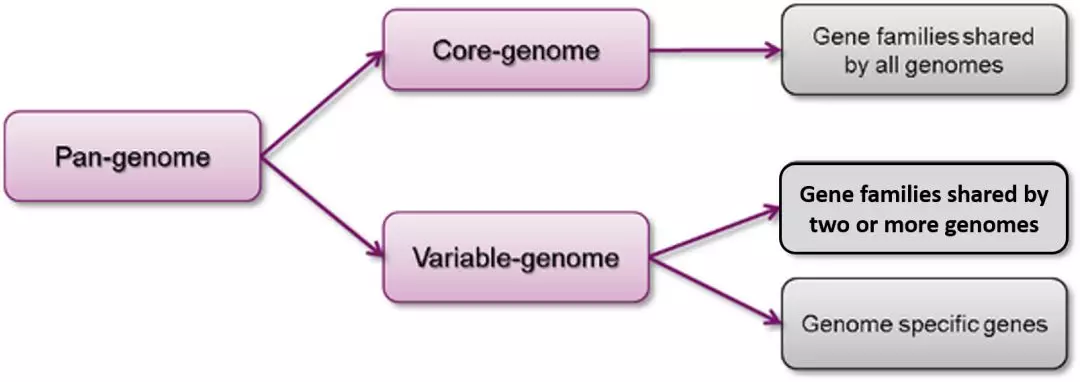
结构变异中的存在/缺失变化(PAVs: Present/absent variations)是泛基因组学的重点研究对象。如上图,泛基因组进而可以分为核心基因(core genes)和可变基因 (variable genes)。核心基因指的是,在所有动植物个体或者菌株中都同时存在的基因。可变基因是指,在一个或者一个以上的动植物个体或者菌株中存在的基因,它们不是固定的,而是多变的。如果某个基因,仅存在某一个动植物个体或者菌株中,该基因还可以细分为品系或者菌株特有基因。一般来说,核心基因大多控制着生命体基本新陈代谢的功能,因为它们广泛存在所有个体中,是不可缺少的。可变基因,往往只存在于一部分个体中,可能就是导致个体产生特异性的性状(抗病性,抗寒性等重要农艺性状)的原因。
为什么我们要开展泛基因组研究?
在漫长的进化过程中,由于地域因素,环境因素等的影响,每个个体都形成了极其特别的遗传性状,单一个体的参考基因组已经不能涵盖这个物种的所有遗传信息,换句话来说,就是如果只使用单一的参考基因组进行遗传驯化变异的研究,你可能会丢失掉很多有意义的基因内容,因为很多独特的序列都不在参考基因组上。另外一个原因,由于基因测序的价格变得更加廉价,为近年来火爆的泛基因组的研究提供了可能性。
技术路线
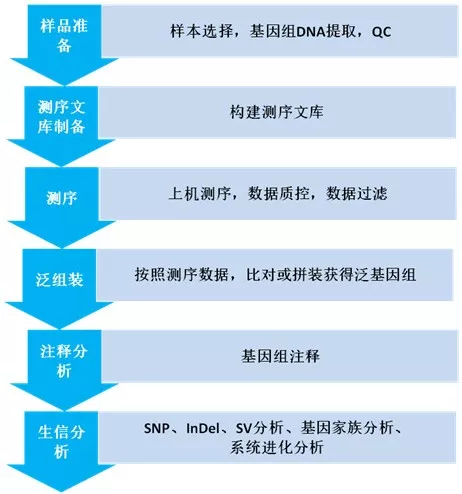
组装技术对比
目前比较流行的研究方法有三种:不基于参考基因组的重头组装,基于参考基因组的迭代组装,以及“map to pan”。下面简单和大家介绍一下这几种方法:
不基于参考基因组的重头组装

这是构建泛基因组或者参考基因组最经典的方法,分别对多个个体分别进行从头组装并且注释,然后将所得的每个个体的组装好的序列与参考序列基因组进行互相比较,找出比对不上的区域或者基因,这些个体独特的基因就是可变基因。该方法不基于参考基因组,可以避免基于基因组方法中由于比对所产生的误差。另外因为是对每个个体进行重头组装,所以该方法可以进一步用于个体之间拷贝数变异(CNVs: Copy number variations)的研究。但是该方法需要比较大的计算资源,需要比较高的测序深度(>50x)或者Mate-pair序列进而增加了预算,以确保重头组装的准确性。这方法需要对每个个体进行组装并且注释,对于含有上百个个体的泛基因组研究不太适合,比如植物基因组的从头组装,这种方法对于小麦这种大型复杂的植物也不太适合。
基于参考基因组的迭代(iterative)组装
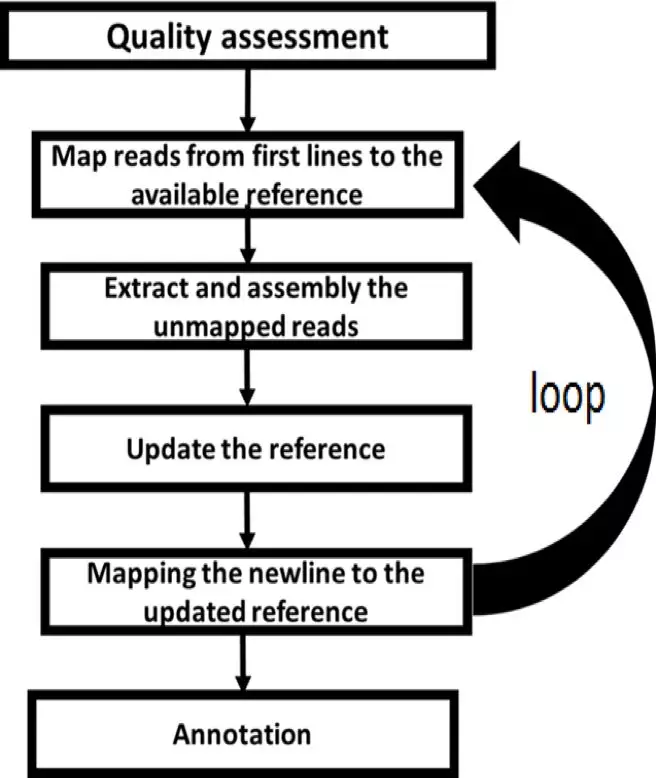
相当于一种迭代的方式,分别将每一个个体的reads先比对到参考基因组中,然后找出没有比对上的部分进行组装,得到新的基因序列进而扩展原有的参考序列。一步一步这样迭代,直到所有的个体都处理完,最后建立起泛基因组,再进行注释。这种方法需要的测序深度较低,因为这种迭代的方法还可以将遗传类型较接近的个体pool在一起进行迭代组装,进而增加组装的深度。另外这种方法因为只是组装没有比对上的部分,相对需要更少电脑资源,比较适合构建群体个体数目比较多的,基因组相对较大的植物基因组。但是这种方法可能会产生更多的小片段,并且无法探究每个个体的CNV。
基于参考基因组的重头组装
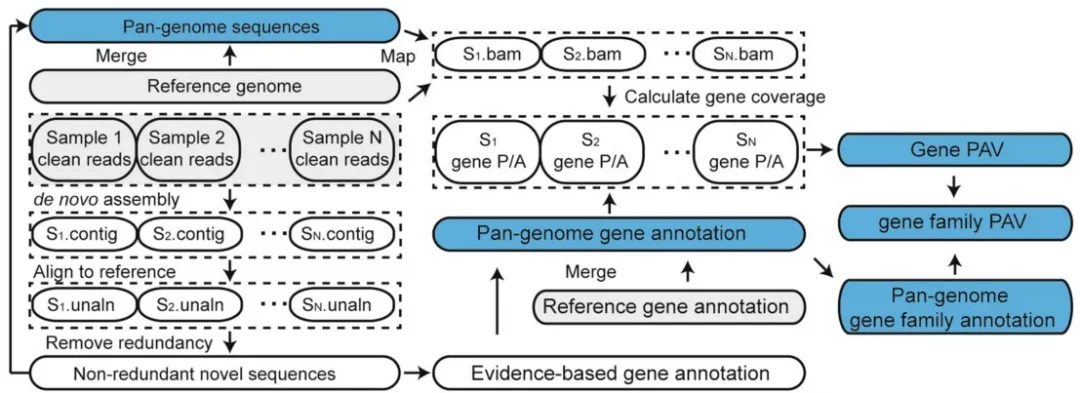
此法又被称为“map to pan”方法,这种方法是先进行de novo组装,然后将组装好的每个个体的基因序列比对到参考基因组中,找出没有比对上的序列,进而将所有没有比对上的序列找出并构建出泛基因组。这种方法也比较适合大规模的群体,但是如果你所研究的物种基因组很大的话,该方法也需要耗费大量的计算机资源。
泛基因组近年来的研究
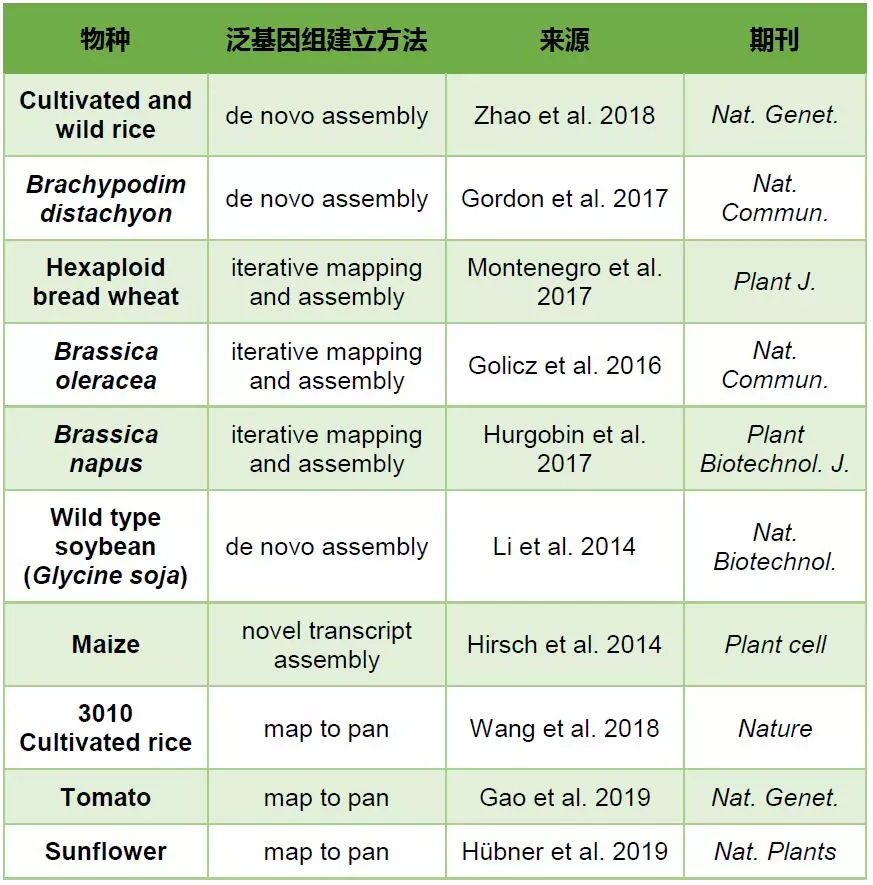
泛基因组的应用
泛基因组测序是运用高通量测序及生物信息分析手段,针对不同亚种/个体材料进行测序及泛基因组组装,构建泛基因组图谱,丰富该物种的遗传信息。泛基因组测序不仅可以获得多个基因组,完善该物种的基因集,还可以获得种群甚至个体特有的DNA序列和功能基因信息,为系统进化分析及功能生物学研究奠定基础。泛基因组也可以看作是对重测序数据的一种挖掘,挖掘发现每一个个体中独特的基因,特别是如果你构建的泛基因组中含有野生种,这能够对野生种的种质资源进行进一步挖掘和分析。
选择不同亚种材料进行泛基因组测序,可以研究物种的起源及演化等重要生物学问题,特别近年来发现PAVs也能看作为其中一种marker,基于PAVs也能对物种进行遗传进化的关联分析;选择野生种和栽培种等不同特性的种质资源进行泛基因组测序,可以发掘重要性状相关的基因资源,为科学育种提供指导;选择不同生态地理类型的种质资源进行泛基因组测序,可以开展物种的适应性进化,外来物种入侵性等热门科学问题的研究,为分子生态学等学科提供新的研究手段。
下期预告
在本次推文主要是介绍泛基因组并且简单的提及了它的应用,但是在目前的植物泛基因组研究中还存在不少的局限性,这里留到第二期来慢慢讲述,敬请大家持续关注。
猜你喜欢
北美华人基因组学在线沙龙(CGM)曾于2018年10月邀请到胡志强博士做了一期泛基因组相关的学术报告。如感兴趣请点击腾讯视频框,收看CGM第 40 期:Towards Eukaryotic Pan-genomics。
参考文献
- Golicz AA, Batley J, Edwards D. 2016. Towards plant pangenomics. Plant Biotechnol J 14:1099–1105.
- Golicz AA, Bayer PE, Barker GC, Edger PP, Kim HR, Martinez PA, Chan CKK, Severn-Ellis A, McCombie WR, Parkin IAP, et al. 2016. The pangenome of an agronomically important crop plant Brassica oleracea. Nat Commun 7:13390.
- Hurgobin B, Edwards D. 2017. SNP Discovery Using a Pangenome: Has the Single Reference Approach Become Obsolete? Biology 6:21.
- Li YH, Zhou G, Ma J, Jiang W, Jin LG, Zhang Z, Guo Y, Zhang J, Sui Y, Zheng L, et al. 2014. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat Biotechnol 32:1045–1052.
- Maretty L, Jensen JM, Petersen B, Sibbesen JA, Liu S, Villesen P, Skov L, Belling K, Theil Have C, Izarzugaza JMG, et al. 2017. Sequencing and de novo assembly of 150 genomes from Denmark as a population reference. Nature 548:87–91.
关注我们
CGM网站:cgmonline.co
YOUTUBE频道:Chinese Genomics Meet-up
腾讯视频频道:Cgmonline 欢迎订阅!
http://v.qq.com/vplus/d4528aec249262f5ea703ce04b40377a
微信群: 请添加微信好友beckyhao13,请务必注明“CGM”。

若有收获,就点个赞吧
0 人点赞
