编辑整理:杨雪蓉、李秋实、郝樾
为从事基因组学研究的华人学者提供一个交流、学习与合作的平台是CGM成立的初衷。经过逾两年半的发展,CGM已经成为一个拥有近700多名华人学者的社群,其中包括来自世界各地的大学教授,研究员、博士后、在校学生,各行各业的科研工作者,大家的专业横跨分子生物学、遗传学、基因组学、生物信息学、生物统计学、进化生物学等不同领域。在CGM微信群中常常会有关于学术问题的精彩讨论。不少群友解决了自己遇到的难题,分享了自己的经验体会,找到了合作的伙伴。为了将这些精彩内容保存下来并分享给更多的华人学者,我们特此推出【CGM微信群精彩问答】这个新专题。因为内容很多而我们能力有限,只能做到挂一漏万。有兴趣的朋友可以根据文末的信息加入CGM微信群,问出你的问题,讲出你的答案。
软件作者发布新版本信息:
区树俊 ISU:大家好,EDTA终于更新了!在@苏维佳-ISU 和我的努力下,EDTA现在支持各种生物基因组的TE注释啦。欢迎试用和反馈👉 https://github.com/oushujun/EDTA 。有问题请直接开Issue,这样后面的人都能受益。多谢大家支持!预印版文章👉 另一个软件LTR retriever也早就available👉 https://github.com/oushujun/LTR_retriever 。测试来测试去还是发现自己家的最好😂。而且EDTA把这整个流程都整合了,不需要自己准备输入文件。
另一个软件LTR retriever也早就available👉 https://github.com/oushujun/LTR_retriever 。测试来测试去还是发现自己家的最好😂。而且EDTA把这整个流程都整合了,不需要自己准备输入文件。
perl EDTA_raw.pl -genome genome.fa -type ltr -threads 36 这就相当于只跑LTR_retriever,简化了很多。
袁道军-ISU-HZAU:👍 安装便利、功能强大、计算高效
李瑞东 Gilead:树俊,用perl开发的软件安装的时候需要提前装好各种module吗?
区树俊 ISU:我没有用bioperl,所用到的modules都是系统自带的,其他的依赖全部用conda安装
恭喜新作发表,与作者交流探讨
杨金良-UNL:Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data (Zhang et al. 2019. Nat Plants 5: 833-845) 恭喜@唐海宝 教授!👏
省伟-NJAU:唐老师,ALLHiC这个流程有没有试过小麦?
唐海宝:@杨金良-UNL 谢谢!@省伟-NJAU 没有试过。一般异源多倍体(如小麦)如果亚基因组差异大,拼接并没有太大问题,就是不同单倍型不会装混。我们这个流程解决的是单倍型序列比较接近的情况(同源多倍体,如甘蔗、苜蓿、土豆)。
区树俊 ISU:有机会试试蓝莓,异源四倍体祖先分化大概1MYA
唐海宝:我也在蓝莓基因组文章上(Colle et al. 2019. GigaScience 8: 1–15),这个应该是Dovetail做的?
区树俊 ISU:🤝才意识到我们是素昧平生的coauthors。ALLHiC能对付二代测序+HiC的情况吗?
唐海宝:看情况,有特定的场景。现在有很多杂合/多倍体基因组拼接错误地将alleles组装在一起,让人觉得是“串联重复”。这种错误用ALLHiC就可以有效地避免。
鲍志贵 OE:要用ALLHiC的话,前期组装必须用Canu一类对杂合和allele敏感的组装软件?
唐海宝:对,要在contig水平把allele单独装出来。一般目前用PB/ONT都自动能办到。
关于GWAS分析
陈雨露-ISU:现在做glm的GWAS大家用的是什么软件或者R包呀
杨金良-UNL :gemma也可以glm吧
省伟-小麦组学:最近有个新的R包rMVP可以做GWAS,挺全的,自动出图等
梁智凯-UNL:刘小磊开发的MVP不错,不用来回整理数据格式
李广伟-河南农大-HENAU:935348004 @陈雨露-ISU 这个是rMVP维护的QQ群,你可以加这个,刘小磊老师和他的一作学生开的群。
谭瑞娟-MSU:Google搜Rtools, 下载安装,然后再装rMVP
关于FST分析
郭婷婷-ISU:求助,我算了基于一个基因全部SNP的Fst,用的是Nei的算法,值是0.42。有没有大神告诉我,这个值是不是太高了。也叫GST。
杨金良-UNL :算diversity的时候,不能只考虑variant sites, invariant sites should also be considered。
崔融丰 MPI-AGE:有一个问题,你算 gst 的时候,是只算至少一个种群里面具有多态性的位点,还是连 fixed 的位点也算?比如,一个种群都是 A, 另一个都是 T , 這种位点有没有算进去啊?好像应该固定了的不同位点也要算吧?那些都是 Fst = 1 啊。
杨金良-UNL :只算有多态性的,那就肯定偏高。要用sequence本身来算。
崔融丰 MPI-AGE:这要看你的两个种群的分化程度。兩个种群开始分化的时候会有很多共有的多态性位点,然后慢慢的就会随机固定,並且各个种群就会慢慢积累自己独有的突变,所以计算時还要考虑那些固定了的不同。物种完全分开后,就没有共享的多态位点了,Fst 就变成 1 了。我觉得只要你计算过程正确,算出來是多少就多少呗 很多時候物种就是分类学家说了算。
郭婷婷-ISU:可不可以这样理解,单个位点都可以算个Fst,我这样算属于多个位点combine averaged Fst。
黄恺驰UBC: 可以的 但单个基因的范围和全基因组水平的分化比还是比较局限。主要还是看你想回答什么问题,0.4这个值没有对比没法说高还是低。
崔融丰 MPI-AGE:是的,单个位点可以算,然后可以多个位点合并。但我记得不是简单的平均。我好像使用的是Weir方法。
区树俊 ISU:可以参考 Huang et al. 2012. _Nature 490: 497–501
郭婷婷-ISU:我也找到一篇文章:Estimating and interpreting FST: The impact of rare variants(Bhatia et al. 2013 Genome Res 23: 1514-1521)
前方高能!! 学习资源大分享
- Part 1 数量遗传学
宗宇OSU_ZJNU:我最近也在估算Fst,是种群间的。用的是ANGSD,两个种之间的差异,有些高值到了0.7甚至0.9。计算方法是基于Fumagalli et al. 2013 _Genetics 195: 979–992。对算法和公式一脸茫然😳。
杨金良-UNL :可以看一下Graham Coop的popgen notes (https://gcbias.org/population-genetics-notes/)。对数量遗传感兴趣的,参见**本人的一些课件**(https://jyanglab.com/AGRO-931-2018/)。从Falconer&Mackay的第八章开始,Statistical Foundation。
区树俊 ISU:谢谢分享!工作量好大啊,能否分享一下怎么准备的课程?
杨金良-UNL :我给自己挖了一个坑,哼哧哼哧填上的。。。写书编教材技术层面建议Github,Overleaf,CSS, Rmd, R package: xaringan 等等先了解一下。内容方面的话,我上过这门课,简化了参考书。
- Part2 生信可视化R代码
李瑞东 Gilead :分享下最近整理的常用的生物信息数据可视化的R代码。方便大家画图时使用。浏览器中打开 https://rli012.github.io/BioinfoHub-pages/book/ 即可。目前收录的比较偏下游。转录组分析用到的可能更多些。基因组分析等还没总结,欢迎指正和多多交流!
编者补充:常用R包ggtree画系统发生树的朋友,可以参考余光创Y叔的教材 https://yulab-smu.github.io/treedata-book/
生信分析标准化流程
杨金良-UNL :我们建CGM还有一个初衷,目前尚未实现:就是把基因组的可视化脚本、或其他常规流程标准化起来。
吴昕-GrandOmics&NextOmics:建议编流程使用WDL/CWL规范,不要重新发明轮子。
孟浩巍 PKU:我觉得Snakemake也很好用呀。我自己录过一系列免费教程视频 https://ke.qq.com/course/393358
同步文件、记笔记的工具
樊维姝-UNL-KIB:请问不同设备之间文件同步,有什么推荐的工具吗?我目前用的是坚果云,但有数据限制。
庄轩-UC:我一直用Dropbox很多年了。
刘科辉-SYSU:百度云,勉强顶用,还免费。
鲍志贵 OE:OneDrive也还不错。
赵飞-sibs:GoodSync可以学习一下。
雷栗_JGI:我用OneNote,现在同步很好,无论什么平台,只要有网就行。
省伟-NJAU:印象笔记也不错嘛,手机端、电脑端都支持,它的剪藏功能很赞,支持收藏微信的文章。
何逸建-RAI:Evernote比较适合第一时间快速记录琐碎的信息,所谓第二大脑。而OneNote很接近真实的笔记,更适合思考后整理笔记。两者可以互补。
祁新帅BHB、省伟-NJAU:推荐一下Typora,好用的很。尤其图片插入功能,配合在线图床使用很方便。
邹枨-Cornell:我用Typora记工作流程,Evernote记各种杂事,用Zim管理本地文件和资源。
添加引用工具全指南
刘科辉-SYSU:各位,我发现Endnote添加reference,会有各种小细节的格式错误,如何破解?
陆俊豪-university Alberta:识别有错误很正常,都要手工矫正的啊。。
陈林兴-UCB:感觉Google Docs的插件还是挺好用的。
沈星星@VanderbiltU:可以自己写一个脚本,根据doi自动从NCBI抓下来,然后想怎么format都行。我博士的时候用过Mendeley,也可以根据doi或PubMed id自动同步提取。
杨金良-UNL :LaTeX + Google Scholar,再也不用担心文献引用的问题。
区树俊 ISU:Endnote,Paperpile,还有其他,都会有各种格式小错的。Robin Buell教给我的方法是,每当发现citation错误,一定要回到软件或数据库里修改,这样只需要改一次,下次再用就没问题了,永远不要在文章里修改。
余光创_SMU_bioinfo:Linux系统下可以用LibreOffice + Zotero。
LaTeX入门
刘科辉-SYSU:有没有推荐入门的LaTeX软件?
邹枨-Cornell,魏馨竹 UCB:Overleaf 可以online写,也有很多template。
崔融丰 MPI-AGE:使用LaTeX好处是什么呢?
李勤UBC:排版便利、非常规范。对数学公式友好。如果用过markdown的话,上手不会太难。
杨金良-UNL :LaTeX是必须要有的啊!尤其是你的ms上需要一个红酒杯印的时候:
或者你想发Nature,review偏偏不接受的时候: 或者你想要一篇ms in preparation format的时候!
或者你想要一篇ms in preparation format的时候!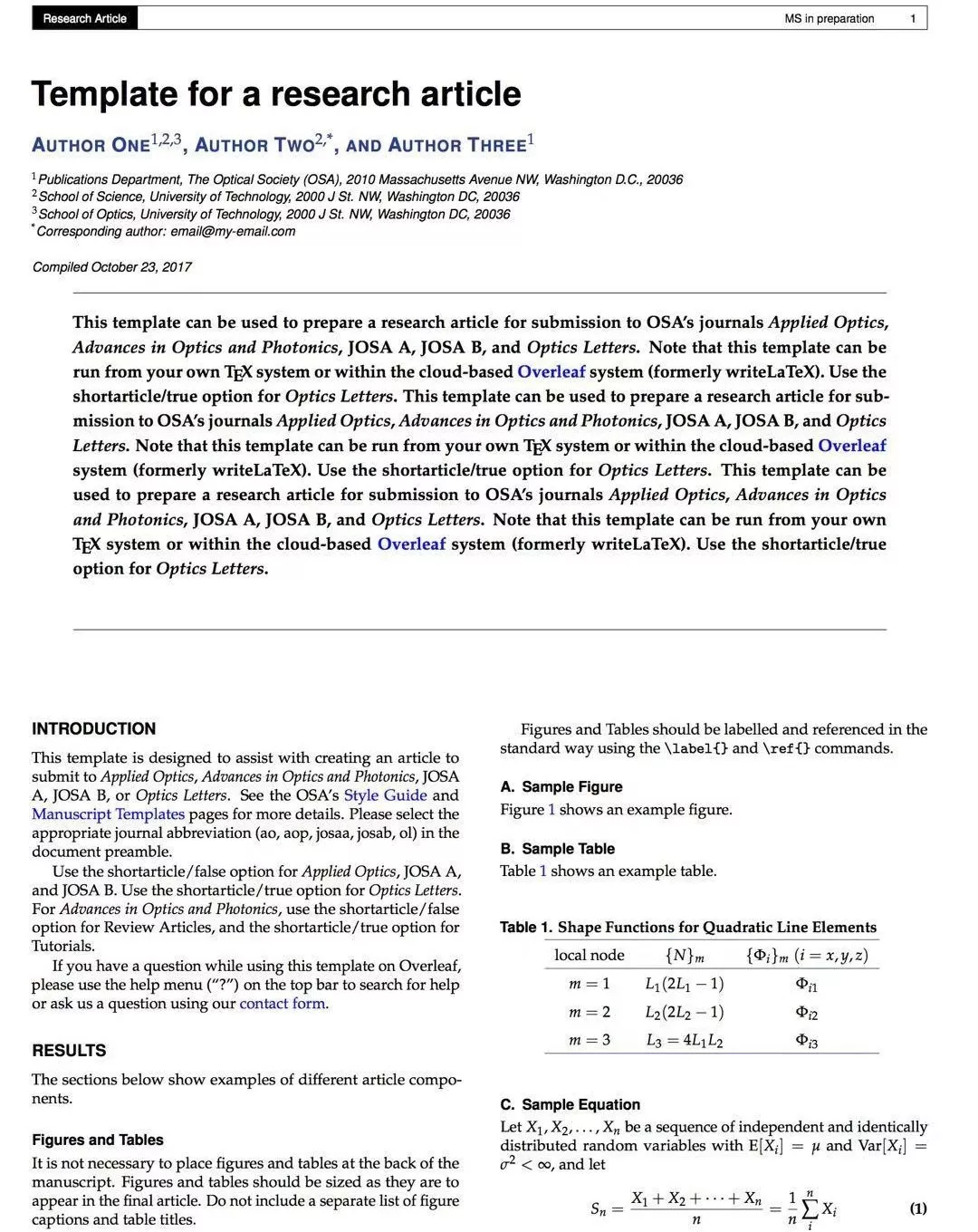 或者你想很短时间搞定一个poster的时候!
或者你想很短时间搞定一个poster的时候!
我以前写过一个教程:http://yangjl.com/en/codes/2016/09/09/atom 具体操作是Atom+LaTeX+很多add-on。
我现在正在做poster,顺便科普一下:
- 在Overleaf上拷贝一个模板
- Git clone到本地。(也可以在overleaf上直接操作,但少了很多灵活性)
- 用Atom编辑器进行编辑,可以看到实时渲染结果。
你甚至可以看到这个文档,在历史上任何时间,谁做过什么操作。然后可以瞬时回到过去的那个版本。
这些都是跟谢益辉老师学的,特颁发证书,以资证明:)
编者注:谢老师的报告荣获CGM年度最佳视频奖
谢老师的网站:https://yihui.name/ 。谢老师提出了JournalDown Project,用了JournalDown,我们可以解脱于格式的桎梏,只需专心创造内容。

若有收获,就点个赞吧
0 人点赞
