背景
在使用过程中存在一些疑问,所以就认真一探究竟。Normalization 的方法很多,适应的条件也不一样。下面两篇讲的比较清晰建议下看下。
参考:
https://www.jianshu.com/p/a9d5065f82a6
https://www.jianshu.com/p/a3b78bd49bcc
R 包edgeR中calcNormFactors() 函数默认使用的方法为 “TMM”,使用于没有经过其他处理的原始 RNA-seq counts 数据。
原理
参考原作者的文章 A scaling normalization method for differential expression analysis of RNA-seq data. Mark D Robinson and Alicia Oshlack
根据经验,作者提出了一个假设——个体之间大部分的基因表达水平是没有太大变化的,变化的只是少数。一般标准化(包括TPM),都会除以 library 大小,即除以所有基因reads总和,来消除不同批次间测序深度造成的影响。例如下面的,sample A /4,sample B /8,所有基因表达量都一样,是没有差异的。
| gene1 | gene2 | gene2 | gene4 | |
|---|---|---|---|---|
| sample A | 1 | 1 | 1 | 1 |
| sample B | 2 | 2 | 2 | 2 |
但是如果像下面这样,简简单单除以 library 大小是不行的。因为此时 library 大小不同不仅仅受到可能的测序批次的影响,而且受到 差异基因gene4 表达量的影响。sample A /4,sample B /14,你会发现所有的基因都有差异,这是不合理的。
| gene1 | gene2 | gene2 | gene4 | |
|---|---|---|---|---|
| sample A | 1 | 1 | 1 | 1 |
| sample B | 2 | 2 | 2 | 8 |
所以,根据最开始提出的假设,大多数基因是不发生变化的,对 library 大小进行矫正。默认情况下,TMM会修剪Mg值中最高和最低的30%,剩余的基因计算factors。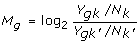
使用
如果只是edgeR 来计算差异基因,很简单。函数返回每个样本library 大小和 factors,不需要其他额外操作可以进行下一步。
library(edgeR)y <- DGEList(counts=rna_seq, group = group)y <- calcNormFactors(y)y$samples## 'data.frame': 262 obs. of 3 variables:## $ group : Factor w/ 2 levels "C","T": 2 1 2 1 1 2 1 1 2 1 ...## $ lib.size : num 41179418 36284427 51657521 43691607 ##37586474 ...## $ norm.factors: num 0.93 0.887 1.01 1.063 0.963 ...
如果我们想自己做些额外的事情,比如我想做生存分析,根据某个基因表达量将群体分为高表达组和低表达组此时可向下面那样做进行简单标准化。
t(t(y$counts) / y$samples$lib.size / y$samples$norm.factors) * 1000000
或者
edgeR::cpm(TMM,log = F) # CPM(Counts of exon model per million mapped reads)
上面两种方法是等价的。相比于TPM,此方法没有考虑转录本本身长度的影响,所以样本内不同转录本丰度我们是无法比较的(即同一个样本内测序为相同reads数的两个转录本,并不表示丰度一样,因为他们本身的基因长度不一样。理论上,基因长的,能检测到的可能性越大,如果reads数相当,表明它表达量少。)。但做差异分析的时候,我们比较的是样本之间的关系,所以这也是为什么edgeR等R包要求输入的是最原始的counts数据而不是TPM吧。(FPKM/RPKM等就不讨论了,已经被很多人认为是错误的。)

若有收获,就点个赞吧
0 人点赞
