质控 Tools for QC
plotCorrelation
此工具基于multiBamSummary或multiBigwigSummary的输出文件。主要用于同一批样本之间的相关性比较,查看是否差异很大,也可做聚类图,来观察哪些样本相似。需求不大,所以先不讲。
plotFingerprint
此工具主要用于Chip-seq数据,通过比较input和实验组观察实验质量。因为Chip实验有高达90%的假阳性。
$ deepTools2.0/bin/plotFingerprint \-b testFiles/*bam \--labels H3K27me3 H3K4me1 H3K4me3 H3K9me3 input \--minMappingQuality 30 \--skipZeros \--region 19 --numberOfSamples 50000 \-T "Fingerprints of different samples" \--plotFile fingerprints.png \--outRawCounts fingerprints.tab
——numberOfSamples 参数主要是随机从样本中抽取的箱数来计算相对的覆盖reads数,-bs 可以设置每个箱的base数。
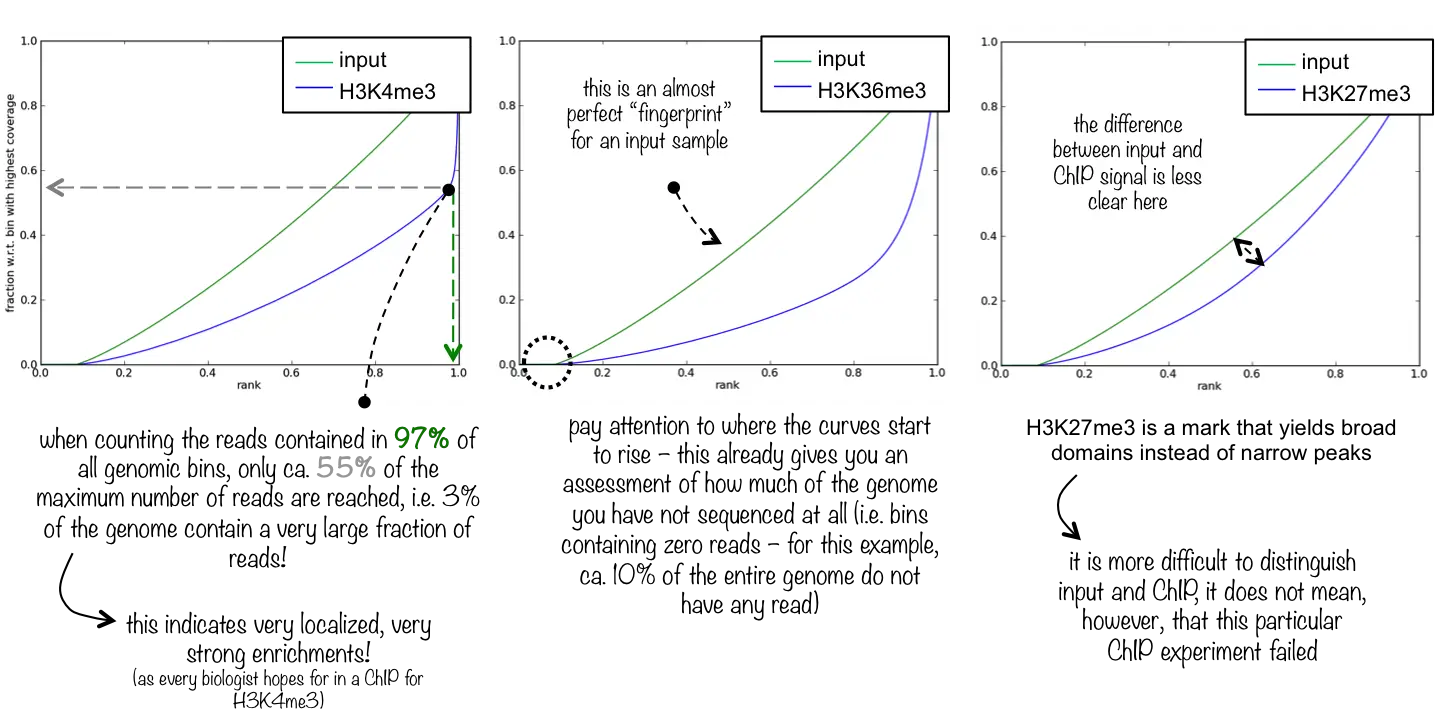
做出的图如上图,大概有三种情况。横坐标是按照bin里read数排序,纵坐标代表bin里reads数的总和百分比。第一个图中,在排序前97%的bin中所有reads数只占了55%,所以有3%的bin中包含了45%的reads。也就是说H3K4me3在某些地方强烈富集,正是我们想看到的。第二个图也很美,而第三个图很难区分input和实验组有什么区别,是个失败的实验。
plotCoverage
该工具可用于评估给定样品的测序深度。它采样100万bp,计算重叠读数的数量,并可以报告一个直方图,告诉你有多少碱基被覆盖多少次。接受多个BAM文件,但它们都应该对应于相同的基因组组装。
$ plotCoverage -b H3K4Me1.bam H3K4Me3.bam H3K27Me3.bam H3K9Me3.bam--plotFile example_coverage-n 1000000--plotTitle "example_coverage" \--outRawCounts coverage.tab \--ignoreDuplicates \--minMappingQuality 10 \--region 19
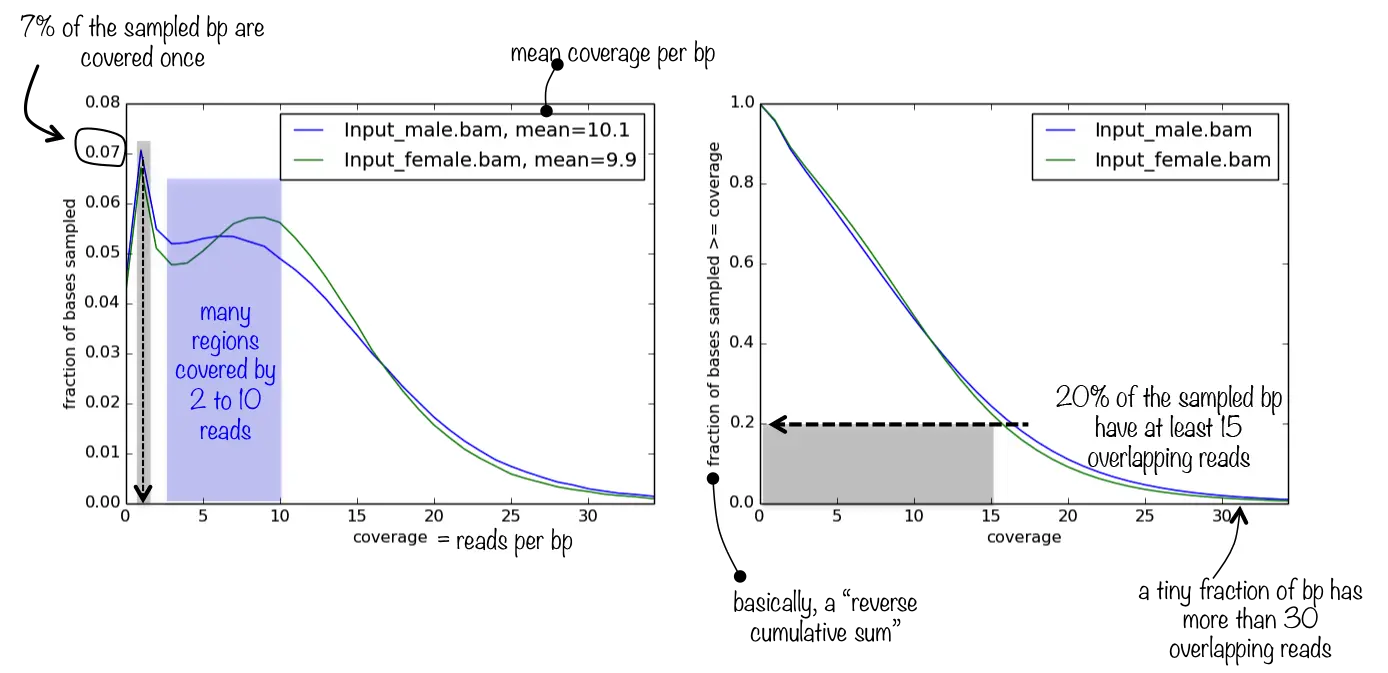
此工具可以看两个样本的覆盖深度相关性。
bamPEFragmentSize
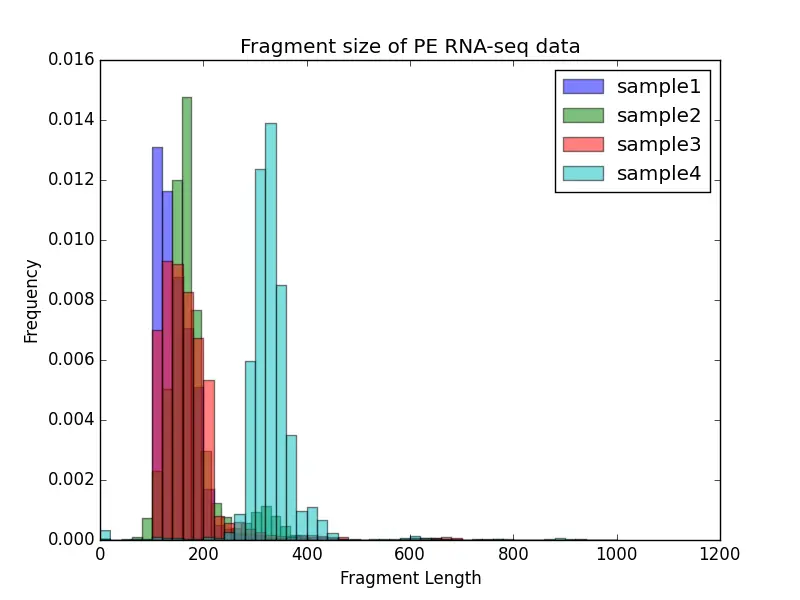
此工具主要估计片段长度和频率。
computeGCBias
计算GC偏差。

若有收获,就点个赞吧
0 人点赞
