一直疑惑MACS2的原理和使用方法,在看了多篇介绍后更是乱的一团糟,最后还是看了官方文档才理清楚。https://github.com/taoliu/MACS/
原理
MACS2的主要功能是 Peak Calling,就是reads富集的正确位置。
其中难理解的就是双峰模型,如下图。在单端测序的时候,如果测序较短,这个时候就会形成双峰,而这双峰并不是真正的蛋白富集的位置。MACS2是基于模型(泊松分布)的方法进行检峰的,意图中的模型是双峰模型,目的是为了将比对上的Reads朝3`端偏移(shift),以更准确地得到蛋白-DNA互作的位置。更详细原理可移步 https://www.jianshu.com/p/0c272643f88b
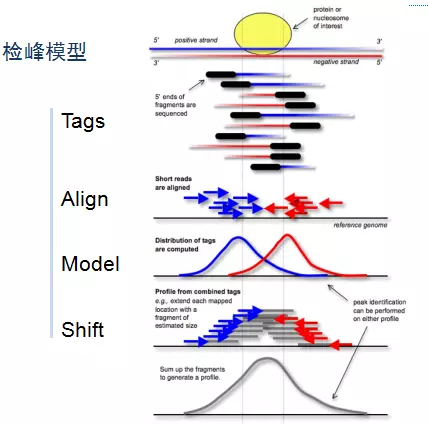
问题
这时我们就遇到一个问题了,上面说的是单端测序的时候,可是我们现在大多数都会使用双端测序。此工具有个 -f 参数。
-f/—format FORMAT
Format of tag file, can be “ELAND”, “BED”, “ELANDMULTI”, “ELANDEXPORT”, “ELANDMULTIPET” (for pair-end tags), “SAM”, “BAM”, “BOWTIE”, “BAMPE” or “BEDPE”. Default is “AUTO” which will allow MACS to decide the format automatically. “AUTO” is also usefule when you combine different formats of files. Note that MACS can’t detect “BAMPE” or “BEDPE” format with “AUTO”, and you have to implicitly specify the format for “BAMPE” and “BEDPE”.
官方说了,此工具可以自动识别大部分输入文件的格式,但是无法区分出”BAMPE” 和 “BEDPE”,也就是无法识别是否是双端测序数据。我们使用“BAM”参数,即使是双端测序结果,软件也只保留 5’ tag的 reads。如果使用参数 “BAMPE” ,将跳过建立双峰模型,根据实际的插入大小来构建峰。同时参数 —nomodel,—extsize 失效。
特殊参数含义
- —extsize : 延长reads 到特定长度,5’ >3’,用这个的前提是要知道蛋白结合的DNA的具体长度,例如核糖体蛋白一周环绕的DNA为173bp,则 —extsize 173 。且只有在—nomodel和—fix-bimodal 生效时使用,而双端测序数据是可以预测read具体长度的,所以不适用此参数。
- —shift: 这个参数是绝对的偏移值,会先于—extsize前对read进行整体移动。
疑惑
如果是nucleosome-seq数据:因为一个核小体大概有147bp DNA缠绕,于是就需要用半个核小体长度进行堆积(pipleup)用于小波分析。参数为—nomodel —shift 37 —extsize 73.
上面是官方文档里的描述,认为nucleosome-seq数据应做此处理,我觉得这也是针对单端测序数据的,如果正负链reads分别延伸到73bp,则两峰之间距离约为73bp,也就是d=73,所以shift=d/2=37。不知道这样理解是否正确。
参考
https://github.com/taoliu/MACS/
https://www.jianshu.com/p/0c272643f88b
https://www.jianshu.com/p/6a975f0ea65a

若有收获,就点个赞吧
0 人点赞
