背景
查看某些基因在特定癌症里是不是发生了拷贝数变异。
最简单的方法
直接去一些分析网站上查看,当然也有一些弊端,例如数据更新滞后,没有给出明确的分析方法和判断标准等,心中不踏实。
- TCGA Copy Number Portal(http://portals.broadinstitute.org/tcga/home)这个网站直接可以查找特定癌症里某些基因是发生了扩增(Amplifications) 还是删除 Deletions,以及q值和突变频率。
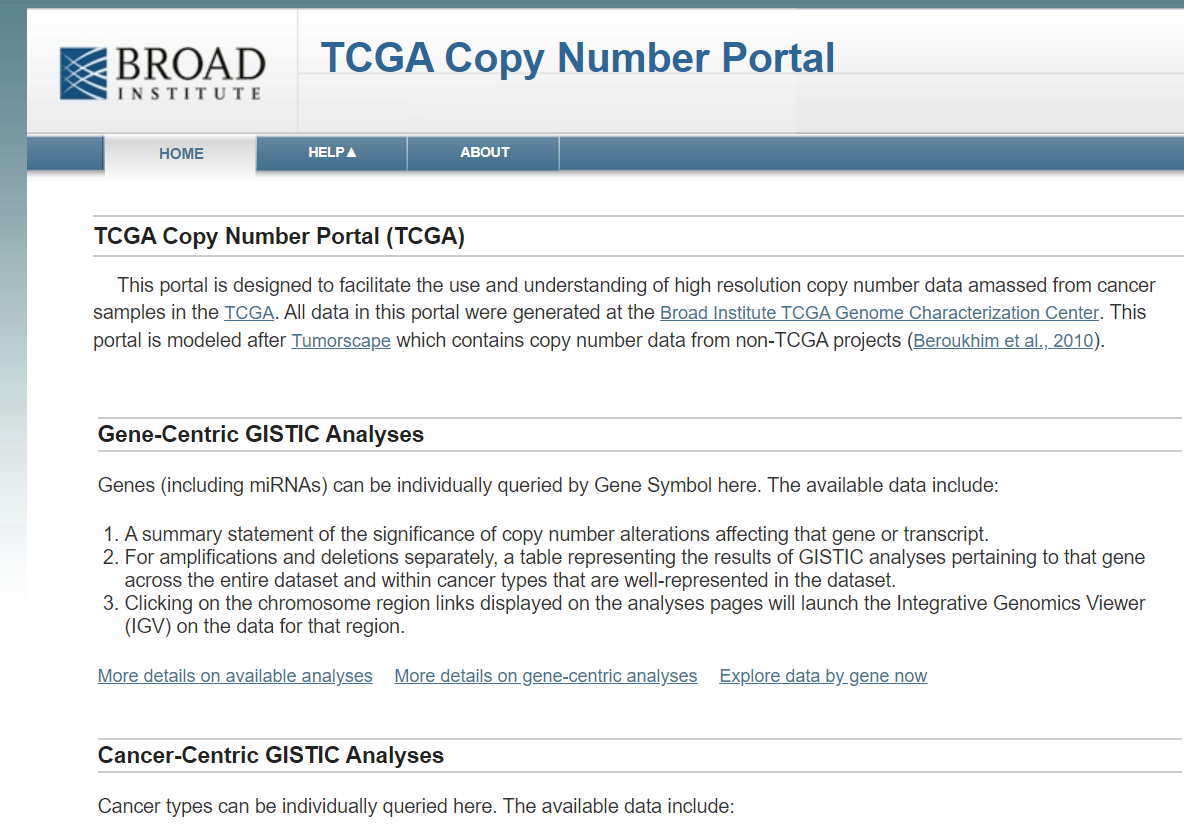
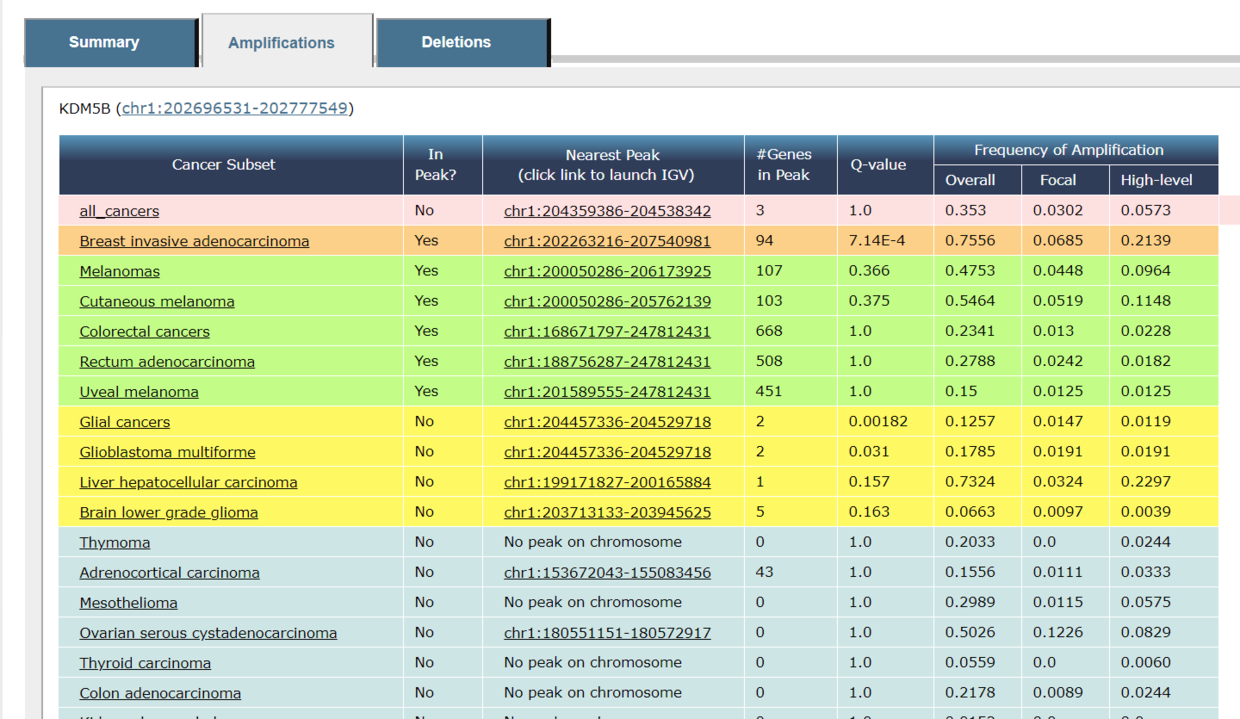
- CbioPortal( https://www.cbioportal.org/) 这个网站中也能找到TCGA某个癌症中某个基因的扩增和删除以及频率。但是查看所有数据后会发现一个基因是在不同个体中是可以扩增也可以是删除的,合理但是也会困惑。同时这个网站也没办法告诉你扩增和删除的程度是多少。所以如果只是初步查看是可以的,但实际来分析数据做出漂亮的图是不行的。
TCGA 基本流程和文件
- TCGA CNV pipeline . TCGA的CNV数据都是来自于 Affymetrix SNP 6.0 array。首先是使用 DNAcopy 进行了处理(暂时没时间,还不清楚方法和原理,也觉得没必要从头开始,除非是处理最原始的数据),得到一个基因区间和此区间的拷贝数的表(Copy Number Segmentation),如下共6列。最后一列Segment_Mean值就是 log2(copy_number/ 2), 正常来说人是二倍体生物则此value值为0,如果拷贝数小于2(删除)则小于0,拷贝数大于2(扩增)则大于0.

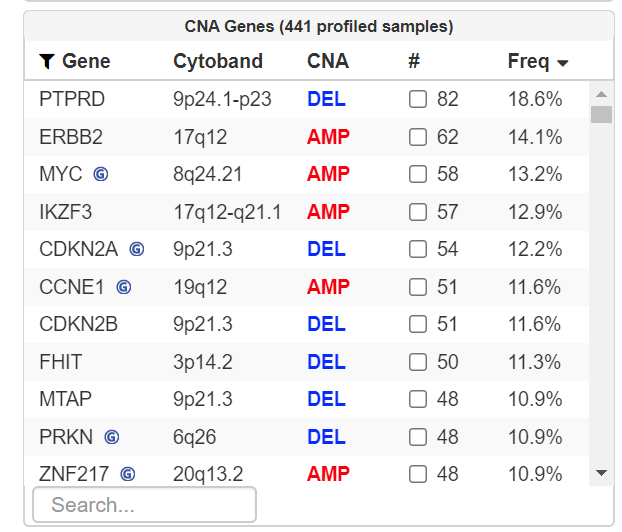
- 此数据可以通过多种途径下载,但是还是要注意一些坑的。1) 最好的方法是直接去TCGA下载,更新的快,但看到443个病例有918个拷贝数变异数据。。。因为里边基本有一半是正常样本的数据不是肿瘤组织数据。2) 其次可以去 https://xenabrowser.net/ 下载将多个样本合并好的数据,但是少了Num_probes这一列,对后续分析还是有一定影响的。 3) 使用http://gdac.broadinstitute.org/ 下载整合好的数据,但是数据很古老停留在了2016年,而且参考基因组是hg19,前面两个网站使用的是hg38。
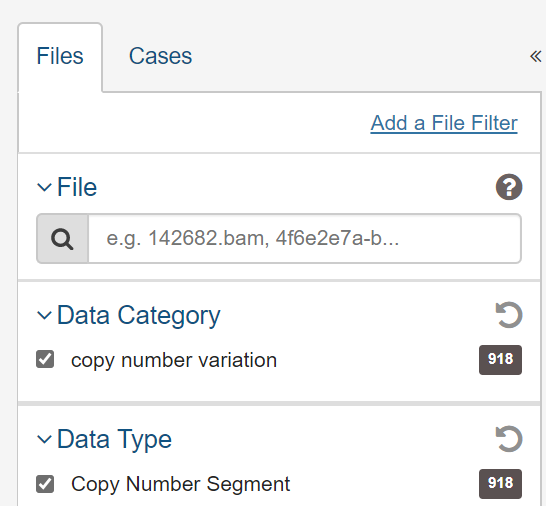
- 另一个相似的数据是 Masked Copy Number Segment, 此表是在上面数据上过滤掉了一些与生殖和性染色体相关的数据。
- 最后一个相关的表是 Copy Number Estimation ,是将序列区间聚焦到了基因上并评估基因是否是扩增或者缺失的。此数据是使用软件 GISTIC2 对 Masked Copy Number Segment进行处理后结果文件focal_data_by_genes.txt 的进一步加工,小于-0.3的值记作-1,大于0.3记作1,-0.3~0.3记作0.
- 在GDC中此表格叫做 Gene Level Copy Number Scores,并且是汇总好的。
拷贝数变异显著性检验
处理CNV数据的一个挑战是我们不知道某个基因或者片段的拷贝数改变是随机的还是在特定表型(癌症)中重复出现,需要一些统计方法来检验显著性,当然样本越多越好。如果感兴趣可以查看相关的文章:
- Detecting independent and recurrent copy number aberrationsusing interval graphs
- TCGA Workflow: Analyze cancer genomics and epigenomics data using Bioconductor packages
- GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers
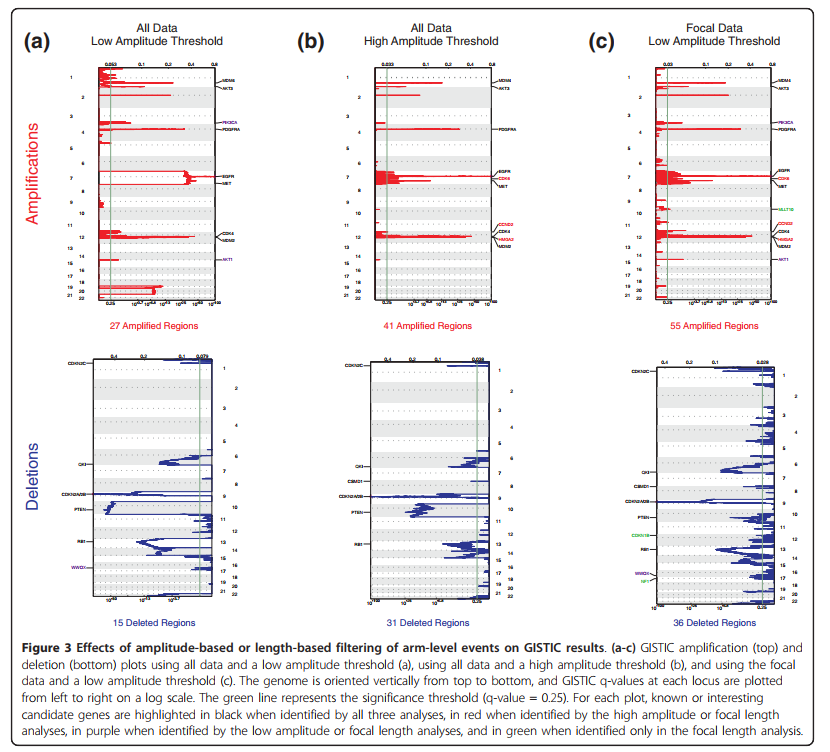
- Mutational Landscape of the Essential Autophagy Gene BECN1 in Human Cancers
总结下,分别使用了RAIG、GAIA、GISTIC2、Fisher exact test 方法,大概只有第四种最简单了吧。。但好像也是依赖于GISTIC处理的。
我们就使用TCGA官方使用的GISTIC2.0 软件吧,虽然官方也用这个软件做了处理给出了我们一个阉割版的focal_data_by_genes.txt 文件,GISTIC2结果文件是很多的,在使用 R包 vaftools 进行作图的时候需要多个结果文件作为输入。
GISTIC2使用
我没有下载安装这个软件,所以不介绍安装方法。但是有更好的解决方法,在 Genepattern(https://cloud.genepattern.org/) 云分析平台中有GISTIC2模块,可以直接注册使用,缺点是上传数据有点慢。详细使用方法请查看 ftp://ftp.broadinstitute.org/pub/GISTIC2.0/GISTICDocumentation_standalone.htm

输入文件需要:
- Segmentation File,这个前面已经介绍过,使用TCGA Masked Copy Number Segment 文件。使用R语言下载整理例子如下,主要是选择”Masked Copy Number Segment”并只保留肿瘤样本(”01A”)。
library(dplyr)library(TCGAbiolinks)query <- GDCquery(project = "TCGA-STAD",data.category = "Copy Number Variation",data.type = "Masked Copy Number Segment")GDCdownload(query, method = "api", files.per.chunk = 100)segment_dat <- GDCprepare(query = query)segment_dat$Sample <- substring(segment_dat$Sample,1,16)segment_dat <- grep("01A$",segment_dat$Sample) %>%segment_dat[.,]segment_dat[,1] <- segment_dat$Samplesegment_dat <- segment_dat[,-7]write.table(segment_dat,"MaskedCopyNumberSegment.txt",sep="\t",quote = F,col.names = F,row.names = F)
- Markers File,下载地址 https://gdc.cancer.gov/about-data/data-harmonization-and-generation/gdc-reference-files ,选择文件 SNP6 GRCh38 Remapped Probeset File for Copy Number Variation Analysis,并注意提示 If you are using Masked Copy Number Segment for GISTIC analysis, please only keep probesets with freqcnv = FALSE ,所以只保留 freqcnv = FALSE 的行和前三列。
- Reference Genome File,此文件云平台中直接有,但要选择好是hg19还是hg38。
来自TCGA官方的参数:
gistic2
-b
-seg
-mk
-refgene
-ta 0.1
-armpeel 1
-brlen 0.7
-cap 1.5
-conf 0.99
-td 0.1
-genegistic 1
-gcm extreme
-js 4
-maxseg 2000
-qvt 0.25
-rx 0
-savegene 1
(-broad 1)
输出文件:
del_qplot.png
all_data_by_genes.txt
focal_data_by_genes.txt
all_lesions.conf_95.txt
gistic_inputs.mat
all_thresholded.by_genes.txt
raw_copy_number.pdf
amp_genes.conf_95.txt
raw_copy_number.png
amp_qplot.pdf
regions_track.conf_95.bed
amp_qplot.png
sample_cutoffs.txt
broad_data_by_genes.txt
sample_seg_counts.txt
del_genes.conf_95.txt
scores.gistic
del_qplot.pdf
结果文件作为 maftool 包输入,根据需要可以做多个图,如下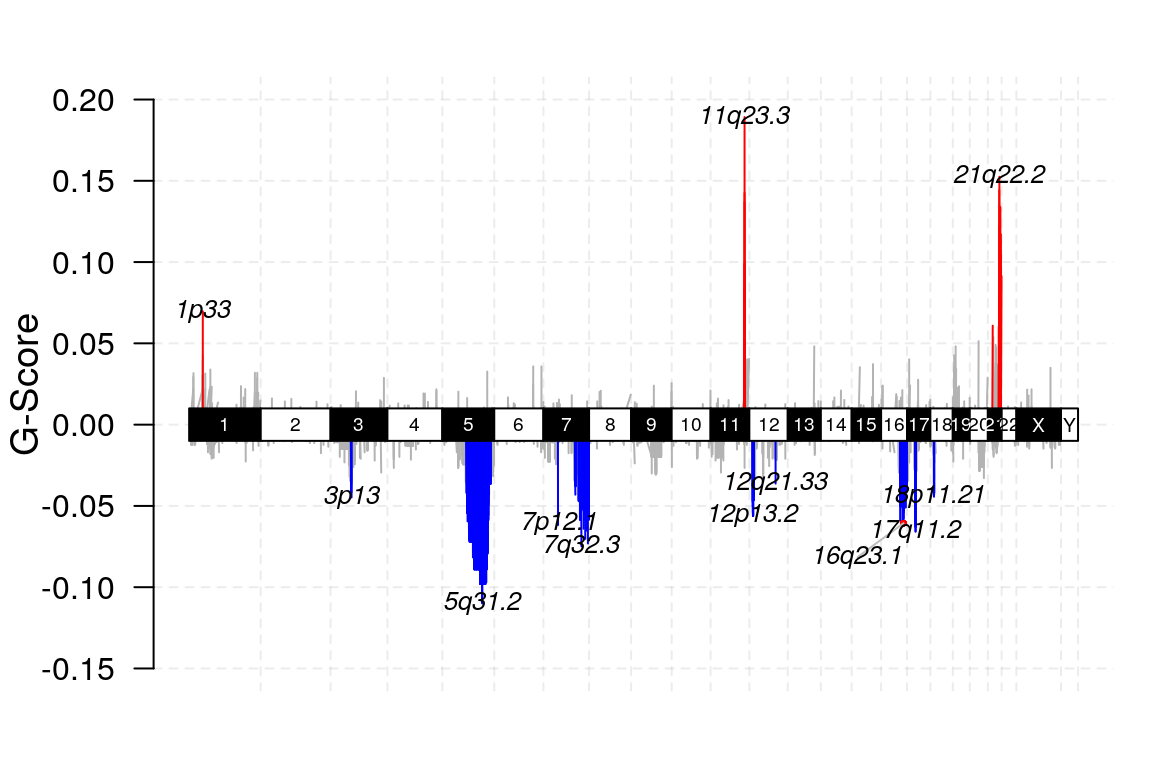
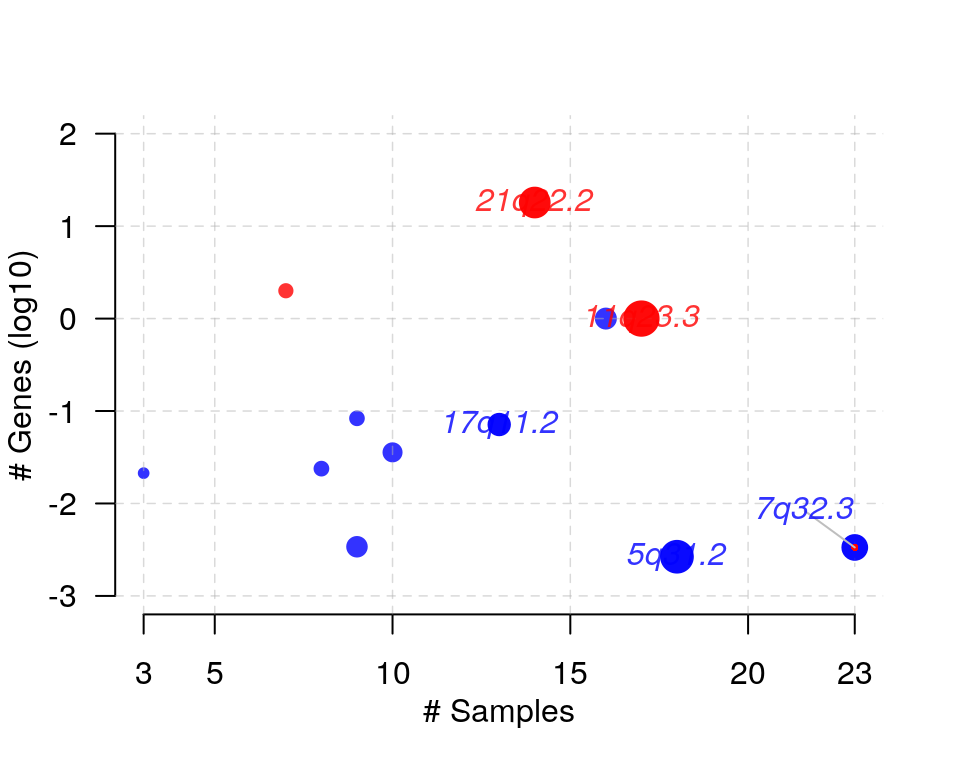
当然你也可以根据自己需要使用RCircos包可视化等。


若有收获,就点个赞吧
0 人点赞
