从GEO下载数据
下载地址https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE121424
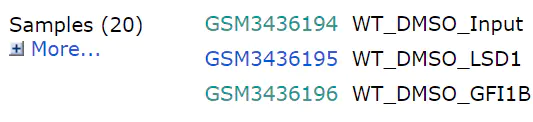
以第一个样本GSM3436194为例,点进链接后最下面有SRA链接(第二张图 ARX4901015),再点进链接,看到SRR8073138编号。

下载之前我们先写个文件记录要下载的文件与编号的对应关系,后面用的到。文件格式如下
(base) [longfei@localhost GSE121424]$ cat infoSRR8073138 WT_DMSO_InputSRR8073141 WT_DMSO_H3K4ME2SRR8073142 WT_DMSO_H3K27ACSRR8073143 WT_GSK_InputSRR8073146 WT_GSK_H3K4ME2SRR8073147 WT_GSK_H3K27AC
下载工具有多个,利用用R包GEOquery,使用aspera软件等。我们用最简单的方法,ncbi官方软件SRA Toolkit,请自行安装。
$ cat info | while read -a line;do (nohup prefetch ${line[0]} -O sra &);done
根据SRR编号循环后台下载到新建的sra文件夹中。
ls sraSRR8073138.sraSRR8073141.sraSRR8073142.sraSRR8073143.sraSRR8073146.sraSRR8073147.sra
提取fastq文件
同样使用SRA Toolkit 工具,提取到fq 文件夹中。
cat info | while read -a line;do (nohup fastq-dump --gzip --split-3 -A srr/${line[0]}.sra -O fq &);done
ls fqSRR8073138.sra_1.fastq.gz SRR8073141.sra_1.fastq.gz SRR8073142.sra_1.fastq.gz SRR8073143.sra_1.fastq.gz SRR8073146.sra_1.fastq.gz SRR8073147.sra_1.fastq.gzSRR8073138.sra_2.fastq.gz SRR8073141.sra_2.fastq.gz SRR8073142.sra_2.fastq.gz SRR8073143.sra_2.fastq.gz SRR8073146.sra_2.fastq.gz SRR8073147.sra_2.fastq.gzSRR8073138.sra.fastq.gz SRR8073141.sra.fastq.gz SRR8073142.sra.fastq.gz SRR8073143.sra.fastq.gz SRR8073146.sra.fastq.gz SRR8073147.sra.fastq.gz
fastqc 质控
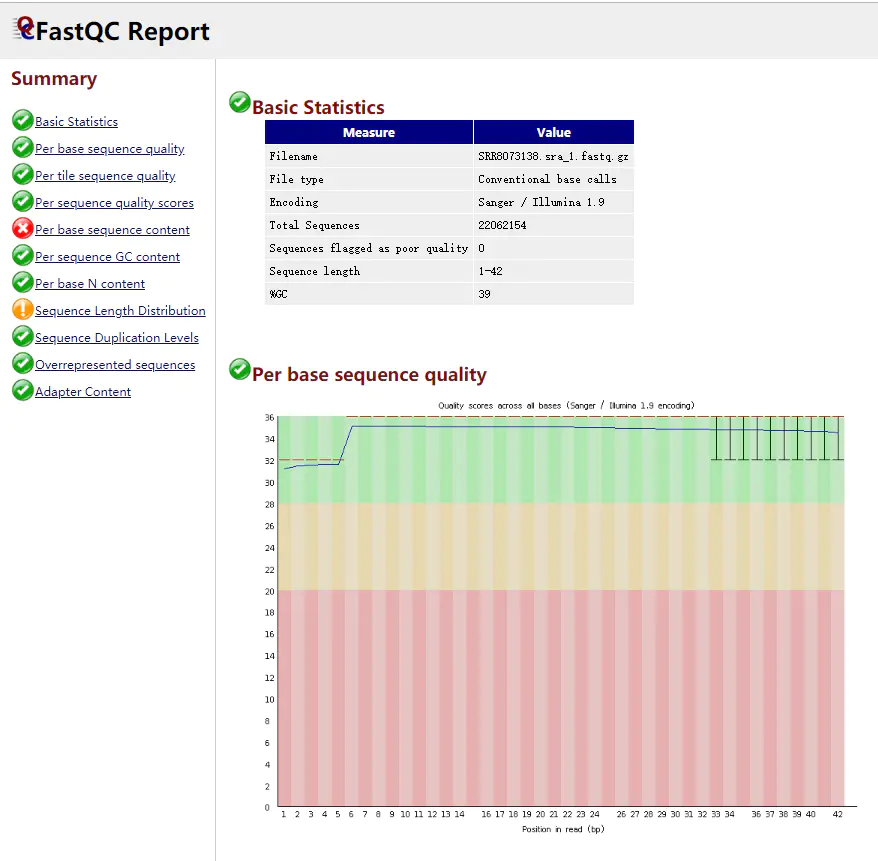
我只挑了其中的一个fastq文件看了下,一般都是处理好的,包括去接头和去除低质量的碱基。
fastqc fq/SRR8073138.sra_1.fastq.gz -o ./ --noextract
生成一个html文件和一个压缩包,传到自己的电脑上,打开html检查质量。
SRR8073138.sra_1_fastqc.html SRR8073138.sra_1_fastqc.zip
将fastq文件比对到基因组上
此处使用软件bowtie2,提前下载好参考基因组文件,这里用hg19。
bowtie_ref=homo_ref/bowtie/hg19mkdir samcat info | while read -a line;do ( nohup bowtie2 -x $bowtie_ref -p 4 -1 fq/${line[0]}.sra_1.fastq.gz -2 fq/${line[0]}.sra_2.fastq.gz -S sam/${line[1]}.sam & );done
将生成的sam文件通过排序变成bam文件,使用软件samtools。这两步可以通过管道合并成一步,省去生成sam文件占用空间浪费时间,但是我经常在这里遇到问题,所以分成两步。
mkdir bamls sam |while read sam;do (nohup samtools sort -O BAM -@ 4 -o bam/${sam%.*}.bam sam/$sam & );done
去除重复序列
使用软件Sambamba,可以查看我的另一篇文章Sambamba 去除重复工具。
mkdir rmdupls bam | while read bam;do sambamba markdup -r -t 4 bam/${bam} rmdup/${bam%.*}.rmdup.bam ;done
使用 bamTools 工具质控
详细命令含义及结果含义移步https://www.jianshu.com/p/2fddb062c503
主要是看ChiP是否合格,想要的地方是否富集到了,区别于开头的测序数据质控。
plotCoverage
查看测序深度相关性
mkdir qcplotCoverage -b rmdup/*bam \--plotFile qc \-n 10000000 \--plotTitle "example_coverage" \--outRawCounts coverage.tab \--ignoreDuplicates \--minMappingQuality 10
plotFingerprint
比较input和实验组,如果差距不大,说明的富集的不好。
plotFingerprint \-b testFiles/*bam \--labels H3K27me3 H3K4me1 H3K4me3 H3K9me3 input \--minMappingQuality 30 \--skipZeros \--region 19 --numberOfSamples 50000 \-T "Fingerprints of different samples" \--plotFile fingerprints.png \--outRawCounts fingerprints.tab
—numberOfSamples 参数主要是随机从样本中抽取的箱数来计算相对的覆盖度
使用 deepTools 工具生成bw文件可视化
详细命令含义及结果含义移步 https://www.jianshu.com/p/e7e2c65183fd
bamCoverage
此命令用于单个样本标准化生成bw文件,使得样本之间可以互相比较。bw文件可用IGV软件可视化。
mkdir bwls rmdup/ | grep -v bai | while read bam ;do \(nohup bamCoverage --bam rmdup/$bam \-o bw/${bam%*rmdup}.bw \--binSize 10 \--normalizeUsing RPGC \--effectiveGenomeSize 2685511504 &);done
computeMatrix
将多个样本放入一个矩阵中,输入为上面生成的bw文件,为后续可视化作图做准备,如plotHeatmap,plotProfile作图。
mkdir resultnohup computeMatrix reference-point \--referencePoint TSS \-b 3000 -a 10000 \-R homo_ref/hg19.bed \-S bw/H3K4Me2_DMSO_a.bw bw/H3K4Me2_DMSO_b.bw bw/H3K4Me2_OG86_a.bw bw/H3K4Me2_OG86_b.bw \-o result/H3K4me2_TSS.gz \--outFileSortedRegions result/H3K4ME2_gene.bed \--skipZeros \-p 10 &
plotProfile
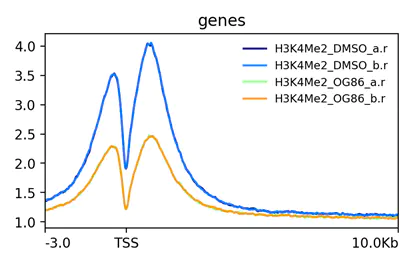
使用plotProfile生成gene在TSS区的富集图
nohup plotProfile -m H3K4me2_TSS.gz -out H3K4me2_TSS.png --perGroup &
使用 MACS2 call peak
参考
https://github.com/taoliu/MACS/
https://www.jianshu.com/p/0c272643f88b
https://www.jianshu.com/p/6a975f0ea65a
nohup macs2 callpeak -t rmdup/H3K9me2K.rmdup.bam -c rmdup/Input-K.rmdup.bam -f BAMPE -g hs -n H3K9me2K -q 0.05 --nomodel &

若有收获,就点个赞吧
0 人点赞
