第一章 绪论
这一章将为你介绍这门课的主讲老师,介绍这门课的学习内容,介绍什么是生物信息学,还有将和你一起学习这门课的小明同学。
首先介绍这门课的主讲老师,巩晶,来自山东大学基础医学院,座机:0531-88380202,手 机 : 1870531** ( 请 和 老 师 索 取 ), 微 信 公 众 号 : CRC_BIOINFO , 工 作 邮 箱 :gongjing@sdu.edu.cn。巩晶老师从中国海洋大学拿到海洋生物学学士学位后,去了德国。先学习了一年德语,之后在德国慕尼黑大学和慕尼黑工业大学联合创办的生物信息学专业取得了本硕连读的硕士学位。之后又用了三年时间在慕尼黑大学取得了生物信息学博士学位。目前的主要研究方向包括蛋白质三维结构的预测、分析及动力学模拟、虚拟筛选、生物数据库、数据挖掘、以及系统发生学等。如要想要了解更多正在开展的研究项目,可以前往课题组网站一探究竟:http://www.crc.sdu.edu.cn/bioinfo/。博士学习期间巩晶老师做过德国巴伐利亚州超算中心的兼职科研助理,主要从事一个统计学语言程序包的平行计算开发。2010 年毕业后进入山东大学基础医学院工作至今。截至目前以第一作者及通讯作者身份发表 SCI 论文6 篇,累计影响因子 17,累计获得科研经费 68 万元。
自 2010 年,山东大学基础医学院正式开设生物信息学课程。首先开设的是研究生全英文课程,截止目前已开设四届,选课学生以留学生为主。2013 年泰山学堂生命取向为大三年级的同学开设了本课程以及配套的实验课程,共计 120 学时的必修课,已开设五届。同年7 月本课程获得山东大学校级精品课程称号。山东大学基础医学院本科阶段和研究生阶段的专业选修课分别于 2015 和 2016 年陆续正式开课。2017 年 9 月山东大学校内课程与中国大学MOOC 线上课程同步开设。
接下来介绍一下这门的考核方法和标准。最终的成绩由三部分组成:单元测验(30%),单元作业(50%),和期末考试(20%)。单元测验都是选择题和判断题。单元作业是需要动动手实际操练一下的。期末考试也是选择题和判断题,但是光盯着试卷是做不出答案的,仍然需要动动手。最终成绩在 60 到 85 分可以获得合格证书,85 分以上可以获得优秀证书。这门课没有专门的教材,不过这里给你推荐了几本参考教材和资料。首推浙江大学陈铭教授的生物信息学这本书,本课程组的老师都是这本书的编委。还有人卫出版社的生物信息学,科学出版社中译本的生物信息学,清华大学许忠能教授的生物信息学,中科院赵国屏教授的生物信息学。此外,两本英文教材也不错,Bioinformatics for dummies 通俗易懂,Bioinformatics an introduction 严谨全面。如果想要了解这门学科最新的研究进展,可以订阅Bioinformatics 和 BMC Bioinformatics 这两个 SCI 期刊。
普通高等教育“十二五”规划教材: 生物信息学(第二版),陈铭 ,2015,科学出版社
全国高等学校教材: 生物信息学(第二版),李霞,2015,人民卫生出版社
生物信息学(中译本),T. Charlie Hodgman,2013,科学出版社
生物信息学,许忠能 ,2008,清华大学生出版社
生物信息学,赵国屏,2017,科学出版社
Bioinformatics For Dummies, 2nd Edition, Jean-Michel Claverie, 2007, Wiley
Bioinformatics - An Introduction, 2nd Edition, Jeremy Ramsden, 2009, Springer
期刊:Bioinformatics, Impact factor: 5.5, Oxford University Press
期刊:BMC Bioinformatics, Impact factor: 2.8, BioMed Central
探索生物信息学神秘岛-01
这一节我们一起来探索生物信息学神秘岛,看看生物信息学是怎么产生的。
翻开字典,我们会发现非常多 bio 开头的单词,土豆前面加上 bio 就成了有机土豆。化学加上 bio 是生物化学,还有生物测量学,生物物理学,生物数学,以及生化危机,生物恐怖主义。好像所有单词前面都能加上个 bio,于是有人就在信息学前面加上了 bio,有了生物信息学。可是这个单词到底是什么意思呢?
这里有很多人试图对这个词做出解释。但他们都如同瞎子摸象一般,只摸到了生物信息学的一个角落。这些人中有生物医药领域的科研工作者,有制药公司的首席科学家,有开发生物数据库的软件工程师,也有正在为 DNA 检测头疼的警察,还有被转基因所困惑的家庭主妇,有你有我有他,还有很多很多人。大家都在生活和工作中或多或少的涉及生物信息学,但都不是生物信息学的全部。因此我们说,生物信息学是一个交叉学科,涉及各个领域,遍布每个角落。
为了更好的理解什么是生物信息学,我们回顾一下历史,看看生物信息学的起源。很早的时候没有生物信息学,但是已经有了生物学和计算机科学,随着这两个学科的发展,他们俩慢慢的很自然的就走到了一起。
这里我们将生物学的起点定在 19 世纪的拉马克,将计算机科学的起点定在 17 世纪的帕斯卡。拉马克的“用进废退”和“获得性遗传”理论大家应该并不陌生。他主张经常使用的器官会发达,不使用的会退化,比如鼹鼠整天呆在地底下挖洞,所以它的眼睛比较瞎,哺乳动物比如猫因为要吃老鼠,所以就长出了尖牙利齿,而鸟因为是将食物直接吞下不用嚼,所以嘴里没有牙。而且拉马克认为这些后天获得的性状是可以遗传给下一代的。虽然拉马克主义后来被达尔文主义所取代,但是这并不能磨灭他的伟大。他是第一个提出进化理论的人!是生物学伟大的奠基人之一!是进化论的倡导者和先驱!
计算机这边的开场人物是帕斯卡。提到帕斯卡,就要提三件事:压强,三角和计算机。压强单位”帕”就是为了纪念法国物理学家帕斯卡而命名。至于帕斯卡三角在咱们中国叫做杨辉三角或贾宪三角。而且,帕斯卡三角比杨辉三角晚了近 400 年,比贾宪三角晚了近 600 年。最后,为什么说计算机这边要从帕斯卡开场呢,是因为 19 岁的帕斯卡发明了人类有史以来第一台机械计算机。
我们再往前迈一步。生物学这边拉马克之后就是达尔文。计算机科学这边帕斯卡之后是莱布尼茨。达尔文《物种起源》一书中提出的进化论观点,相信你已经背的滚瓜烂熟了,似乎从小学就在讲优胜略汰,适者生存,自然选择,不可抗拒。更有伟大的马克思主义创始人之一恩格斯将”进化论”列为十九世纪自然科学的三大发现之一,对人类有杰出的贡献!莱布尼茨是德国最重要的数学家、物理学家、历史学家和哲学家,和牛顿同为微积分的创建人。他的研究成果还遍及力学、逻辑学、化学、地理学、植物学、动物学、解剖学、地质学、航海学、气体学、语言学、法学、外交学等等。他还是最早研究中国文化和中国哲学的德国人。并且,他在中国八卦图的影响和启发下发明了二进制。如果让阳爻为 1,阴爻为0,那么八卦中的坤,艮,坎,巽,震,离,兑,乾,就对应二进制中的 000,001,010,011,100,101,110,111。莱布尼茨认为:数理逻辑、数学和计算机三者均出于一个统一的目的,即人的思维过程的演算化、计算机化、以至于在计算机上实现。所以说莱布尼茨是首次提出
“计算机”这个概念的人。
再继续往前走,孟德尔三大遗传定律出现让生物学向前迈出了一大步,而莫尔斯发明的电报让信息传播进入了新纪元。早在 1866 年孟德尔就已经提出了遗传因子、显性性状和隐性性状等重要概念,并阐明其遗传规律。但是直到 34 年后,也就是孟德尔去世 16 年之后,孟德尔定律才被世人认可。造成这一结果的原因一是孟德尔没有高水平论文的发表。二是他遇人不淑,择友不慎。内阁里让孟德尔拿山柳菊验证,可那个时候大家不知道山柳菊是无性生殖的植物!今天我们说孟德尔是遗传学的奠基人,现代遗传学之父。
与孟德尔同一时期的莫尔斯,于 1844 年在美国国会议事厅里发出了世界上第一封电报。1858 年,横跨大西洋连接欧美两洲的海底电缆铺设成功。到清光绪年间,有线电报进入中国。可以说有线电报的发明是人类历史上信息传递的一次飞跃。
探索生物信息学神秘岛-02
我们继续探索生物信息学神秘岛。上一次讲到了孟德尔和莫尔斯。在孟德尔之后,生物学的发展突飞猛进。从瑞士人米歇尔分离出核酸,到丹麦的约翰森首次提出基因一词,再到美国的摩尔根在果蝇中发现染色体,只经历的短短的半个世纪。摩尔根因创立了染色体遗传理论而获得了 1933 年的诺贝尔奖,是现代实验生物学的奠基人!到 20 世纪中,生物学和计算机科学又同时放光。
生物学这边,在基因,染色体这些新词汇面世之后不久,美国的三位科学家通过实验证明基因和染色体是由 DNA 构成的,DNA 是生物的遗传物质。同一时期,计算机科学出现了一位天才,图灵。他是英国著名的数学家和逻辑学家,被称为计算机科学之父、人工智能之父,是计算机逻辑的奠基者。在图灵之前没有任何人清楚地说明过莱布尼兹所说的“计算机”到底是怎么一回事。直到 1936 年,图灵向伦敦权威的数学杂志投了一篇题为“论数字计算在决断难题中的应用”的论文,提出著名的“图灵机”的设想。图灵机被公认为现代计算机的原型。图灵后半生遭受迫害,不堪其辱,咬了一口被氰化物浸泡过的苹果,自杀身亡。许多年后,有一位图灵的粉丝,捡起了那个被图灵咬了一口的苹果,创建了著名的苹果公司。
之后 1944 年,美国人 Chargaff 提出了 A=T,G=C 的 Chargaff 规则。同一时期,世界上第一台现代电子计算机“埃尼阿克”,诞生于美国宾夕法尼亚大学。埃尼阿克占地面积 170平方米、重达 30 吨。他的“绝招”是在 1 秒钟内进行 5000 次加法运算。1951 年英国生物化学家桑格将胰岛素的氨基酸序列完整地定序出来,同时证明蛋白质具有明确构造,即,氨基酸像一条链一样排列,同一种蛋白质总是具有特定的氨基酸排列顺序。这项研究使他单独获得了 1958 年的诺贝尔化学奖。
到了 1953 年不得不提的是沃森,DNA 之父。他和克里克于 1953 年在《自然》上发表了 DNA 双螺旋结构模型。而这个模型是根据弗兰克林和弗兰克林的助手威尔金斯,于 1952年用 X 射线衍射法获得的一张晶体照片构建的。因此 DNA 双螺旋结构的发现这四个人都功不可没。然而 1962 年的诺贝尔奖只授予了三位男士。弗兰克林并未获得诺贝尔奖,因为她在获奖前因癌症去世,而诺贝尔奖只能颁发给活着的人。尽管如此,我们还是应该记住这位伟大的女性。
在 DNA 的结构破解不久,第一个蛋白质的晶体结构也与世人见面。桑格告诉我们氨基酸是按照一定顺序排列的,而这个晶体结构告诉我们,他们们并不是一条线,而是折叠成一个具有特定形状的空间结构。这个晶体结构同样是通过 X 射线衍射法获得的。这门技术的应用,使得研究大分子的结构成为可能。
与沃森一起破解 DNA 双螺旋结构的克里克在 1958 年就提出了中心法则,并于 1970 年在《自然》上的一篇文章中重申。克里克提出的中心法则主要是说 DNA 复制形成 DNA,DNA 转录形成 RNA,RNA 再翻译形成蛋白质。今天我们知道,除了这些,病毒中的 RNA也可以自我复制,RNA 还能逆转录成为 DNA,甚至理论上可实现遗传物质从 DNA 到蛋白质的直接转移。在中心法则提出不久,两位美国科学家破译了全部遗传密码字典的 64 个密码子并解释了密码子如何操控蛋白质合成。他们二人和提出 tRNA 三叶草结构模型的美国化学家霍利分享了 1968 年的诺贝尔奖 。
到了 20 世纪 70 年代,桑格再次登场,研究出一种称为链终止法的技术来测定 DNA 序列,又叫做双去氧终止法或桑格法。这项研究成果后来成为人类基因组计划得以展开的关键技术之一,并使桑格于 1980 年与合作者吉尔伯特获得诺贝尔化学奖。这是桑格第二次获得诺贝尔奖。1975 年真的是不同凡响的一年,这一年桑格发明了桑格测序法,而比尔盖茨于这一年成立了微软公司,更巧的是,乔布斯也于这一年成立了苹果公司。
随着测序技术的出现以及计算机科学的快速发展,1979 年美国洛斯阿拉莫斯实验室建立了 GenBank 数据库,以储存测序产生的数据。三年后,欧洲分子生物学实验室 EMBL 也建立了核酸序列数据库,之后亚洲也有了自己的核酸序列数据库 DDBJ。三大核酸序列数据库于 90 年代初实现资源共享,联合成立国际核苷酸序列数据库。随着三大核酸数据库中数据的迅猛增长,生物信息学日渐成熟,并展露出不可或缺的重要地位。1987 年美籍华人林华安博士首创了 bioinformatics 一词,並发起首屆国际生物信息学系列会议,使得生物信息学一词在世界各地广为沿用。非常有趣的是,bioinformatics 这个词也有着自己的进化史,这个词最早以 compbio 出现,是 compute 和 biology 的缩写,后来发展成 bioinformatique,informatique 这个词源于法语,英语化后变成了 bio 连线 informatics。这个词用了一段时间,直到电子邮件的出现。早期的电子邮件,标题不支持连线,于是去掉连线,有了今天的bioinformatics。
生物信息学是神马
这一节我们来看看生物信息学究竟是神马?
人类基因组计划第一个五年总结报告给出了生物信息学较为完整的定义。报告中说:生物信息学是一门交叉学科,它包含了生物信息的获取、加工、存储、分配、分析、解释等在内的所有方面,它运用数学、计算机科学和生物学的各种工具来阐明和理解大量数据所包含的生物学意义。
此外,各国不同的教科书里关于生物信息学也有不同的定义。比如,美国乔治亚理工大学给生物信息学的定义是:生物信息学是采用数学、统计学和计算机等方法分析生物学、生物化学和生物物理学数据的一门综合性学科。
美国加州大学洛杉矶分校说,生物信息学是对生物信息和生物学系统内在结构的研究, 它将大量系统的生物学数据与数学和计算机科学的分析理论及使用工具联系起来。
浙江大学陈铭教授在他所著的《生物信息学》一书中写到:生物信息学是计算机与信息科学技术运用到生命科学,尤其是分子生物学研究中的交叉学科。如果我们把 HGP 第一个五年报告,美国加州大学的定义,美国乔治亚理工大学的定义,还有浙江大学陈铭教授所说的,综合到一起。他们似乎都在表达同一个意思,那就是“生物
信息学就是用计算机来解决生物问题”。可是老师这么多定义背哪个啊?我们这门课是不需要背定义的!
生物信息学的研究对象非常多,只要有生物学意义的他都研究。如果要细分的话,可以分为核酸,蛋白质,和其他。核酸里包括诸如测序及应用,基因序列注释,基因预测,核酸序列比对,核酸数据库,比较基因组学,宏基因组学,基因进化,RNA 结构预测,等等等等。而蛋白质就更加包罗万象了,除了蛋白质数据库,蛋白质序列比对,还有蛋白质二级三级结构预测,蛋白质相互作用分析,分子动力学模拟,分子对接,蛋白质组学,等等等等。至于其他,凡是不能简单归入核酸或蛋白质的都包括在其他里面,比如代谢网路模建,数据挖掘分析,序列算法开发,计算进化生物学,生物多样性研究,等等等等。我们这门课会挑最基础,最常用的内容给大家以最通俗最实用的方式进行讲解。
这门课学神马
你一定很关心,这门课到底学神马?我们当前所在章节是绪论。绪论之后是生物数据库,序列比较,分子进化与系统发生,蛋白质结构预测与分析,基因组学与蛋白组学,序列算法,统计基础,数据挖掘,编程基础与网页制作。
在第一章绪论里,你需要:认识很多人,理解什么是生物信息学,掌握这门课的学习内容,知道如何取得好成绩。
接下来生物数据库这一章非常有用,首先你需要理解为什么需要生物数据库。然后,你会发现原来世界上有这么多生物数据库,他们大致可以分为几类。之后,你将学会几个重要数据库的使用,包括什么时候使用什么数据库,如何在这些数据库中找到你想要的信息,以及如何解读这些信息。
序列比较是生物信息学研究的重要方法。像不像?像!像不像?像!像不像?不知道!没关系你会比较两个人,我来教你怎么比较两条序列,怎么知道他们谁和谁是一家子的,谁不是。当然,这里面需要点数学。五年报告里不是说了嘛!生物信息是生物学+计算机科学+数学。所以这里让大家体会一下生物信息学真正的研究内容,而不是仅限于应用的研究对象。除了序列比对,你一定也听说过 BLAST 这个词。可是,你真的会玩 BLAST 吗?不用急,这部分内容里你会听到一个关于仓鼠的故事。听完这个故事,各种 BLAST 便会运用自如。
分子进化与系统发生这一章我们主要讲树,当然不是这个树,是这个树!这是系统发生树,他可以帮助我们从分子水平了解蛋白质或基因的进化关系。比如从树上你会发现,虽然河马叫河-马,但是他和鲸鱼的关系比跟马近。如果你很喜欢数学的话,这里又可以大算一番了。当然,作为专业人士我们都是用软件来自动创建系统发生树的。比如,这个 MEGA,非常好用,非常好学,当然,最重要的,免费!我会手把手教你如何用这个软件构建准确的系统发生树。
蛋白质结构预测与分析这一章,会给你带来一定的视觉冲击。我会教给你一个 3D 分子查看器的使用。用它就可以玩转 3D 蛋白质。这部分内容将与课程活动挂钩,也就是每年一届的最美蛋白质评选活动。活动获奖率每年都有提升,只要你积极参与,就有机会获奖哦!老师,这些漂亮的结构是从哪里来滴?可以用仪器解析,不过仪器太贵了,不是我们这门的菜。我们擅长的是用最少的钱办最多的事。所以我将教给大家如何快速省钱的用计算机把一条氨基酸序列变成超酷的 3D 结构。蛋白质经常是拉帮结队团伙作案的,通过分子对接我们可以洞悉他们是喜欢面对面,还是喜欢背靠背的呆在一起。此外,通过分子动力学模拟我们还能还原他们在机体中的动态表现。
基因组学与蛋白组学这一章非常高科技。先为你揭秘神奇的测序技术。从第一代的桑格法到第四代的纳米孔,这个技术到底对我们的生活改变了多少?“不治已病治未病”的基因检测到底是如何实现的呢。此外,我们说核酸能测序,蛋白质能测序吗?答案是能!用质谱仪测。这是我们中心的质谱仪,用它可以做各种各样的蛋白质组学分析,比如氨基酸测序,定量蛋白质组分,鉴定翻译后修饰,确定亚细胞定位,胶联质谱研究蛋白质相互作用,等等等等。如果你有兴趣,可以来我们实验室参观哦~!
序列算法这一章里将给你介绍如何把生物问题转化成数学问题,并高效解决。比如,你会学到如何画一棵后缀树,并利用这棵后缀树快速查找重复序列,还会学到如何通过解决最高分子序列问题找出蛋白质的跨膜区。你还将了解到什么是一个算法的复杂度。这是能让你在有生之年解决问题的关键。
统计基础这部分主要讲贝叶斯定理。这是个超牛的定理,你可以用它算一算为什么喊狼来了喊到第三次就没人来了!此外,虽然整天说假阳性,假阳性,你知道除了假阳性之外,还有真阳性,假阴性,真阴性吗?这些又是怎么算出来的呢?
数据挖掘将带你揭开大数据的面纱,攫取里面的财富。你会发现大数据不只出现在电视新闻里,原来他离我们那么近,每个人都可以掌握你身边的大数据,他们非常有趣,非常有意义,只要你挖掘一下,其乐无穷!怎么挖?用洛阳铲吗?用洛阳铲那是土夫子。我们用傻鸟 weka 挖!Weka 是一个数据挖掘的傻瓜机,你不需要知道它里面的算法黑盒子是怎样的,只需要给他数据,他就给你结果。就这么简单。当然最关键的是,WEKA 完全免费!
最后呢学生物信息学不能不学点编程,咱不能离了 excel 就蒙圈啊。你编的小程序如果想让大家都来用,就还得学点儿网页制作。所以说,每个搞生物信息的其实都是一只程序猿。学这真的有用吗?至少毕业找不到工作可以去做网站啊。另外,这个世界上不是只有窗户,还有企鹅,事实上生物信息学领域更常用的是企鹅。这门课还将你熟悉 linux 操作系统。让你成为真正的生物信息学大咖!
第二章 生物数据库
为什么需要生物数据库
生物数据库是生命科学研究领域中十分重要的资源。我们不讲如何创建生物数据库,我们只讲如何使用数据库,比如什么时候该用什么数据库,如何在数据库中查找想要的信息,以及如何解读这些信息。
首先我们来看看这密密麻麻的是什么玩意儿!这是 DNA 序列,你猜对了吗?确切的说,这是 HIV-one 病毒的整个基因组,包含了 9752 个碱基,编码 9 个基因。差不多一页 A4 纸,正反面打印就可以把这九千多个碱基记录下来。当然,病毒的基因组是很小的。与之相比,我们人的基因组就要大得多了,有多大呢?3 个 G,也就是 30 亿个碱基。如果一页 A4 纸打印 5000 个碱基的话,我们需要打印 60 万页,才能把整个人的基因组打印完。如果 600 页装订一本书,60 万页得装订 1000 本书。这样的一本书厚度大约在 3 厘米左右,那么 1000 本3cm 厚的书摆在一起有 30 米长,比一节和谐号火车的车厢是还要长。这只是一个物种的基因组,目前已经有超过 1000 个物种通过测序技术获得了全部基因组序列。那么就需要至少200 个五层 30 米宽的书架。这相当于两个山东大学趵突泉校区图书馆的藏书。即使我们建一个足够大的图书馆,把所有的基因组天书全都放进去了。可是还有比基因组大好几倍的注释信息呐!没有这些注释,天书无法解读。就算把这些注释也都塞进去,现在给你一条 DNA序列,让你到图书馆里去找找,他是哪个基因组里面的,找的到吗?
所以这些庞大的信息不能放在实体店里,而只能放在线上,放在数据库里。事实上两个图书馆也装不下的基因组序列,一块 3T 的硬盘就搞定了。当然把数据放进硬盘只是完成了数据库的收集工作。收入的数据要能方便的访问,要能根据各种条件进行搜索,当然有变动或有新注释的时候还要能够更新。这样一个存储数据的空间加上一套完善的管理系统才能构成一个完整的数据库系统。因此我们说,生物数据库是被组织起来的大量生物数据,这些数据通过计算机可被方便的访问、管理及更新。
讲到这里,加入一个小插曲。我们一起来熟悉一下这些天文数字单位。这些单位到处都能用到,日常生活中也是,尤其是从千到拍。千 kilo,你应该很熟悉,kilometer,一千米。一千简称 1K。1000 个 K,是 1 兆,简称 1M。1000 个 M 是 1G。1000 个 G 是 1T,1000 个 T是 1P,依次 1000 倍 1000 倍的往上增长。谁是谁的 1000 倍,一定要记清楚,千万不要搞混。
生物数据库的分类
这个世界上,到底有多少生物数据库?很难统计,但是用成百上千这个词儿一点儿也不夸张。著名的学术期刊 Nucleic Acids Research 有一个生物数据库专刊。有点儿规模的数据库都争相在这里发表。包括 Genbank,PDB 等等,都在这里发表更新版本。截止 2015 年底,这个专刊收录的生物数据库累计 1685 个。当然还有一些在其他刊物发表的小型专项数据库。加上这些,目前世界上得有超过 2000 个生物数据库。当然,不是所有数据库都是活的。NAR数据库专刊曾经发文指出,已发表的数据库中有相当数量的数据库在发表之后就死去了。事实上,数据库不是一个可以放在那里保质期无限长的罐头,他需要专业人员不断的管理、维护和更新。这些工作一旦停止,数据库便立刻失去了生命力。所以如果将来有一天,你也有了自己开发的数据库,请善待它,不要轻易抛弃他。
可是这么多数据库,一节课讲不完,甚至一年也讲不完啊!所以这里只挑最重要的,最常用的,以及和大家的专业最密切相关的几个数据库进行讲解。正是因为数据库太多,所以大家特别喜欢给他们分类。不同的教材分类原则不同,也就是没有标准的分类方法。我们这里选取了比较好理解的原则,把生物数据库首先分成三大类。核酸数据库,蛋白质数据库和专用数据库。核酸数据库顾名思义,是与核酸相关的数据库。蛋白质数据库是与蛋白质相关的数据库。而专用数据库是专门针对某一主题的数据库,或者是综合性的数据库,以及无法归入其他两类的数据库。核酸数据库和蛋白质数据库又分为一级和二级。一级数据库存储的是通过各种科学手段得到的最直接的基础数据。比如测序获得的核酸序列,或者 X 射线衍射法等获得的蛋白质三维结构。蛋白质的一级数据库还可以再具体分为蛋白质序列数据库和蛋白质结构数据库。二级数据库是通过对一级数据库的资源进行分析、整理、归纳、注释而构建的具有特殊生物学意义和专门用途的数据库。比如从三大核酸数据库和基因组数据库中提取并加工出的果蝇和蠕虫数据库,再比如根据蛋白质三维结构数据库中的结构信息,分析统计出的蛋白质结构分类数据库 CATH 和 SCOP 等。
文献数据库 PubMed:基本使用
我们先从一个简单,但是非常常用的文献数据库 PUBMED 入手。
事情是这样的。我们的 3D 小人 Jim 拿到了一条老板给的基因序列。老板让他好好了解一下这条序列,因为这是他这一阶段工作的主要研究对象。Jim 很头疼,一堆字母怎么研究啊!这时,他听说有个师兄,上过生物信息学慕课,号称生物信息学专家。于是 Jim 把他的序列交给了这位专家。很快专家告诉他,这条序列看上去像是一个 dUPTase。Jim 很开心,太棒了,原来是 dUTPase 啊!可是转过头想问问专家什么是 dUTPase,专家只丢给他一个话:牛仔很忙,找 PubMed 去!Pubmed 是什么啊?
PubMed 是拥有超过两百六十万生物医学文献的数据库。这些文献来源于 MEDLINE,也就是生物医学文献数据库、生命科学领域学术杂志以及在线的专业书籍。他们大部分提供全文链接。注意,提供的是链接,你有没有权限通过这个链接打开或者下载全文另当别论。不管怎么说,看上去还不错。PubMed 主页上有个索条。不管三七二十一,先把家说的 dUTPase 敲到搜索条里,点搜索。找到了五百多条文献。每个文献有题目,作者,刊物,出版时间等等。如果列出的这些信息还不够,或者无法满足你的要求,你从页面上方设置每个文献是要显示内容、总结、摘要,还是其他。还可以控制每页显示几个文献,以及按照你期望的顺序进行排序。
如果找到的文献太多,一时看不完,可以把他们保存到本地。只要选中你要保存的文献,然后通过发送按钮,选择文件,再选择保存的内容以及顺序,最后点创建文件。这样你选中文章的信息就以纯文本文件的形式保存到本地电脑上了。
初次尝试 PubMed 搜做之后,Jim 突然想起来,老板是和一个美国人一起研究这个dUTPase 的。那个美国人他见过,好像是叫 Abergel。那不如,下一步就查查看这个 Abergel发表的文章。这次他在搜索条中输入 Abergel,搜索!这次一共找到四百多条文献,比刚刚搜 dUTPase 少了一些,但还是挺多的,而且世界上叫 Abergel 的人看来不少,找到的文章也大多和 dUTPase 没有关系。看来这些都不是 Jim 见过的那个 Abergel 写的。
经过这次尝试之后,Jim 想到,如果把 dUTPase 和 Abergel 都写到搜索条里,是不是就能找到 Abergel 写的关于 dUTPase 的文章了呢?试一下!果然,就剩 2 条了。第二篇文章,题目里有 dutpase,作者里有 abergel,有点儿意思,点一下题目,看看详细。Pubmed 提供文献的摘要和全文链接。这里有两个全文链接。其中一个链接的图标上有 free 字样。Jim 很幸运,这篇文章是 open access 的,也就是免费阅读的。两个链接,第一个是期刊提供的全文链接,第二个是 PubMed 中心提供的全文链接。点其中一个链接,就可以在线浏览文献全文了,或者下载全文的 PDF 文件到本地。至此,JIM 总算找到了些许有用的信息。
文献数据库 PubMed:高级搜索
回到 PUBMED 搜索结果页面,在显示内容格式这个下拉菜单里,除了总结,摘要,还有个叫 MEDLINE 的项目。你可以把它简单的理解为数据库中文献记录的内部结构。每条文献都是以这样的内部结构存储在数据库中的。一篇文献的所以信息被分割成小节,每个小节都有自己的索引名,比如 TI 代表题目,AB 代表摘要,AU 代表作者等等。这些由几个字母组成的索引名是规定好的。
了解了 MEDLINE 结构,我们就可以在搜索条中通过引入索引名,来按照不同的规则搜索。比如搜索 Down 这个词。我们在 Down 的后面加上空格,中括号 AU(Down [AU]),就会返回所有作者名里有 Down 这个词的文献。如果加上[TI],则返回题目中有 Down 的文献。中括号 AD 是搜索发表单位。如果什么限制都没有,只写 Down 的话是在任意地方搜索。我们看到,引入不同索引名后,搜索到的文献数量是不相同的。
现在,我们的 Jim 已经学会了 PubMed 的基本使用。他打算趁暑假回北京期间找个相关的专家拜访一下。于是他在 PubMed 上搜索,题目和摘要里有 dUTPase 的文献,而且文献的发表单位是北京的(dUTPase [TIAB] Beijing [AD])。搜索后,找到了这四篇文章。其中第二篇文章是研究 dUTPase 晶体结构的,Jim 很感兴趣。点进去看一下,找到最后一位的作者。我们说,通常课题的主要负责人会以通讯作者的身份出现在作者列队的最后。那么,这个研究团队的主要负责人就是这位 Su XD,Su 教授。他的单位是北京大学。之后的任务就可以交给度娘来完成了。度娘很快就找到了苏教授的地址和电话,而且连照片都有。Jim通过这些信息,成功的拜访了北京大学生命科学学院的苏晓东教授。
除了搜索条,我们还可以利用高级搜索工具更精确的查找。高级搜索可以无限的添加条件。比如发表日期从哪天到哪天,文章类型是 research article 还是 review,语言是英语发表的还是其他语言发表的等等。我们可以查找比如 2000 年至今发表的,题目有 dUTPase 关键词的,英语的,review 文章。一共找到五篇,相信 Jim 同学看完这 5 篇文章之后,一定会对dUPTase 有一个很好的了解。
关于 PubMed 还有几点要说明的。在搜索时,可以使用引号。引号里的词会被当作一个整体来看待,而不会被拆开。这个小技巧同样适用于度娘等网络搜索引擎。也可以使用逻辑词 AND OR NOT。比如你可以规定,题目里有 dUPTase,并且题目里有 bacteria,但是作者里不要有 Smith(dUTPase [TI] AND bacteria [TI] NOT Smith [AU])。此外,如果知名知姓,可以利用正确的名字缩写来提高搜索的准确度。还有非常关键的一点,每篇文章都有自己唯一的 PubMed ID(PMID)。通过这个号,可以直接找到某一篇文章。最后,不得不说的是,有的时候 PubMed 也帮不了你。比如,搜索 1995 年以前的文献中排名十位以后的作者是白费力气。搜索 1976 年以前的文献是没有摘要的。搜索 1965 年以前的文献,就别想了。关于 PubMed 就给大家介绍到这里。
一级核酸数据库:GenBank 原核生物核酸序列(1)
这一节我们来看一级核酸数据库,他主要包括三大核酸数据库和基因组数据库。三大核酸数据库包括 NCBI 的 Genbank,EMBL 的 ENA 和 DDBJ,它们共同构成国际核酸序列数据库。三大核酸数据库,美国一个,欧洲一个,亚洲一个。美国的 Genbank 由美国国家生物技术信息中心 NCBI 开发并负责维护。NCBI 隶属于美国国立卫生研究院 NIH。欧洲核苷酸序列数据集 ENA 由欧洲分子生物学研究室 EMBL 开发并负责维护。亚洲的核酸数据库 DDBJ由位于日本静冈的日本国立遗传学研究所 NIG 开发并负责维护。Genbank,EMBL 与 DDBJ共同构成国际核酸序列数据库合作联盟 INSDC。通过 INSDC,三大核酸数据库的信息每日相互交换,更新汇总。这使得他们几乎在任何时候都享有相同的数据。
我们以 NCBI 的 Genbank 为例,教你如何解读一级核酸数据库。我们将分别浏览一个原核生物的基因和一个真核生物的基因。为此,请先跟我复习一下原核生物与真核生物基因的不同之处。原核生物基因组小,真核生物基因组大。原核生物基因密度高,1000 个碱基里就有 1 个基因,而真核生物基因密度低,比如人,要 10 万个碱基才有 1 个基因。与此对应,原核生物编码区含量高,而真核生物低。此外,原核生物的基因是呈线性分布的,而真核生物的基因是非线性的,因为翻译蛋白质的外显子被内含子分隔开来。也就是真核生物的mRNA 要经历剪切的过程,剪切后的成熟 mRNA 才能进行翻译。这是原核生物和真核生物基因的最大区别,即,原核生物没有内含子,真核生物有内含子。这个巨大的区别,将导致两种基因在数据库中不同的存储及注释方式。
我们首先来看一条原核生物的 DNA 序列,它是编码大肠杆菌 dUTPase 的基因,在Genbank 里的数据库编号是 X01714。从 NCBI 的主页(http://www.ncbi.nlm.nih.gov/)选择Genbank 数据库。Nucleotide 数据库就是 Genbank 数据库,然后在搜索条中直接写入这条序列对应的数据库编号 X01714,点击“搜索”。结果返回编号为 X01714 的序列在 Genbank 中详细记录。从这条记录的标题我们得知,dUTPase 是脱氧尿苷焦磷酸酶,编码他的基因叫dut 基因,所属物种是大肠杆菌。下面是关于这个基因的详细注释,我们逐条浏览一下:
LOCUS 这一行里包括基因座的名字,核酸序列长度,分子的类别,拓扑类型,原核生物的基因拓扑类型都是线性的,最后是更新日期。
DEFINITION 是这条序列的简短定义,也就是前面看到的标题。
ACCESSION 就是在搜索条中输入的那个数据库编号,也叫做检索号,每条记录的检索号在数据库中是唯一且不变的。即使数据提交者改变了数据内容,Accession 也不会变。你会发现,这条记录里,Accession 和 Locus 是一样的。这是因为这个基因在录入数据库之前并没有起名字,因此录入数据的时候便将检索号作为了基因的名字。但是有些基因,在录入数据库之前已经有了自己的名字,那么这些基因所对应的 Accession 和 Locus 就不一样了。你可以这样理解,Locus 是一个同学的真实姓名,而 Accession 是这个同学的学号。同一个人在不同的学校里会有不同的学号,而名字只有一个。基因也是一样,同一个基因在不同的数据库中会有不同的检索号,而基因的名字只有一个。
Version 版本号和 Locus,Accession 长得差不多。版本号的格式是“检索号点上一个数字”。版本号于 1999 年 2 月由三大数据库采纳使用。主要用于识别数据库中一条单一的特定核苷酸序列。在数据库中,如果某条序列发生了改变,即使是单碱基的改变,它的版本号都将增加,而它的 Accession 也就是检索号保持不变。比如,版本号由 U12345.1 变为 U12345.2,而检索号依然是 U12345。版本号后面还有个 GI 号。GI 号与前面的版本号系统是平行运行的。当一条序列改变后,它将被赋予一个新的 GI 号,同时它的版本号将增加。容易混淆的就是 LOCUS,ACCESSION,VERSION 和 GI。后面的都很好理解。
KEYWORDS 提供能够大致描述该条目的几个关键词,可用于数据库搜索。
SOURCE,基因序列所属物种的俗名。他下面还有一个子条目,ORGANISM,是对所属物种更详细的定义,包括他的科学分类。
REFERENCE 是基因序列来源的科学文献。有时一条基因序列的不同片段可能来源于不同的文献,那样的话,就会有很多个 REFERENCE 条目出现。REFERENCE 的子条目包括文献的作者、题目和刊物。刊物下面还包括 PubMed ID 作为其子条目。
COMMENT 是自由撰写的内容,比如致谢,或者是无法归入前面几项的内容。
FEATURES 是非常重要的注释内容,它描述了核酸序列中各个已确定的片段区域,包含很多子条目,比如来源,启动子,核糖体结合位点等等。
source 说明了核酸序列的来源,据此可以容易的分辨出这条序列是来源于克隆载体还是基因组。可以看到,当前序列来源于大肠杆菌的基因组 DNA。
promoter 列出了启动子的位置。细菌有两个启动子区,-35 区和-10 区。-35 区位于第286 个碱基到第 291 个碱基 ,-10 区位于第 310 个碱基到第 316 个碱基。
misc_feature 列出了一些杂项,比如,这条说明了从第 322 个碱基到第 324 个碱基是一个推测的,但无实验证实的转录起始位置。
RBS 是核糖体结合位点的位置。
CDS,Coding Segment,编码区。对于原核生物来讲,CDS 记录了一个开放阅读框,从第 343 个碱基开始的起始密码子 ATG 到第 798 个碱基结束的结束密码子 TAA。除了位置信息,还包括翻译产物的诸多信息。翻译产物蛋白的名字是 dUTPase,这个编码区编码该蛋白的第 1 到第 151 个氨基酸。翻译的起始位置和翻译所使用的密码本,以及计算机使用翻译密码本根据核酸序列翻译出的蛋白质序列。需要强调的是,这不是生物自然翻译的,而是计算机翻译的。事实上,蛋白质数据库中的大多数蛋白质序列都是根据核酸序列由计算机根据翻译密码本自动翻译出来的。中间部分是翻译出的蛋白在各种蛋白质数据库中对应的检索号。
通过这些检索号可以轻松的链接到其他数据库。
此外,X01714 这条核酸序列还包含第二个“潜在的”基因,也就是计算机预测出来的基因。它编码的蛋白目前的数据库里没有详细记录,是个未知的蛋白。像这样,一条核酸序列包含多个基因的情况在 Genbank 里是很常见的。
ORIGIN 作为最后一个条目记录的是核酸序列,并以双斜线作为整条记录的结束符。至此整条记录就浏览完了。
有时你可能会想要保存这条序列,但是直接从这里拷贝,序列里既有空格,又有数字,不是纯序列,手动删除这些又很麻烦。这时,你可以在这条记录的标题下面找到一个叫做FASTA 的链接。点击他,你会获得 FASTA 格式的核酸序列。FASTA 格式是最常用的序列书写格式,他由两部分组成,第一部分就是第一行,以大于号开始。大于号后面接序列的名称或注释。第二部分就是第二行以后的纯序列部分,这部分只能写序列,不能有其他内容,比如空格,注释,行号之类的都不能在序列部分出现。早期的 FASTA 格式要求序列部分每行60 个字母。但这个规定早已被打破,每行 80,或每行 100,都可以。标题下方,除了 FASTA 链接,还有一个图形化链接,点击可以看到 Features 里的注释信息以图形的形式更直观的展示出来。可以看到这条序列包含的两个基因,他们的启动子的位置,核糖体结合位点的位置等。其中一条基因是编码 dUTPase 的 dut 基因,另一个是编码未知蛋白的潜在的通过计算预测出的基因。
如果想要保存这条记录,最好的方法是像保存 PubMed 文献列表那样,点击发送链接,然后选择以纯文本文件的形式保存整条记录到本地电脑上。
一级核酸数据库:GenBank 真核生物核酸序列 mRNA
我们浏览真核生物的核酸序列。真核生物的基因与原核生物不同,是非线性排列的,也
就是基因里有外显子和内含子。因此真核生物核酸序列的数据库记录要要比原核生物复杂。
有时需要几条记录拼凑在一起才能描述出一个完整的基因。我们先来看看编码人 dUTPase
的成熟 mRNA 序列。成熟 mRNA 是已经剪切掉内含子,只剩外显子的序列,所以这条成熟
mRNA 序列和我们之前看到的原核生物的 DNA 序列从拓扑结构上看是几乎一样的,都是线
性的。输入这条成熟 mRNA 序列的检索号 U90223,搜索!
打开数据库记录,基本的注释内容和原核生物的差不多,这里只挑两点特别的地方说一
下。大家看到 KEYWORDS 后面只有一个点。这个点提示我们,数据库并不是完美的,所有
数据库都存在数据不完整的问题。再有,JOURNAL 后面我们看到是写的是未正式发表。但
事实上,这篇文章早在 1997 年就已经发表在 JBC 上了。因此,忠言逆耳:别指望 Genbank
或任何一个数据库能够百分百做到数据无误且实时更新。
Features 里的注释内容与原核生物的数据库记录相似,CDS 指出了从 63 到 821 是一
段编码区,在这段编码区里基因是连续的,因为是经过剪切后的成熟 mRNA,它将被翻译
成线粒体型 dUTPase 蛋白。下面/translation 里给出的是计算机翻译出的该蛋白的序列。
在 Features 里还有两个新的条目之前没有见到过。 sig_peptide 和 mat_peptide。
sig_peptide,也就是 signal peptide,指出了编码信号肽的碱基的位置。信号肽决定了蛋
白质的亚细胞定位,也就是蛋白质工作的地方。mat_peptide,也就是 mature peptide,指
出了编码成熟肽链的碱基的位置。他从信号肽后面开始,到编码区结尾提前三个碱基结束。
编码区一直到第 821 号碱基,而编码成熟蛋白的最后一个碱基是第 818 号碱基,这中间差了
3 个碱基,那最后的这三个碱基干嘛去了呢?编码区的最后三个碱基是终止密码子,不翻译。
这条真核生物序列的 Genbank 注释看起来和原核生物的差不多,这是因为我们很小心的挑
了一条成熟 mRNA 的序列。
一级核酸数据库:GenBank 真核生物核酸序列 DNA
基因组里的 DNA 序列,是非线性分布的基因序列。我们仍然浏览编码人的 dUTPase 的
dut 基因序列。输入检索号 AF018430,搜索!
这个检索号下的序列标题是“人 dut 基因的第三号外显子”。人的 dut 基因肯定包含多
个外显子,而当前的这条 DNA 序列里只包含了一个外显子。其他的外显子在别的数据库记
录里。从 SEGMENT 处可以看到,人的 dut 基因序列被分成了 4 个片段,并且分别存储在 4
条数据库记录中。也就是说,只有把四个片段全部凑在一起,才能拼凑出完整的基因。当前
这条数据库记录是所有四个片段里的第二个。这个片段里只包含一个外显子,是第三号外显
子。需要注意的是,一个片段可以只包含一个外显子,也可以包含不止一个外显子。另外,
这个例子告诉我们,LOCUS 和 ACCESSION 是可以不相同的。
从 FEATURES 里可以找到这个序列片段在染色体上的具体位置。是在 15 号染色体的长
臂上,位置在 15 到 21.1 条带之间。
GENE 这部分指出了拼出完整基因所需的所有四个片段的检索号,以及具体的位置。也
就是 AF018429 这条序列的 1 到 1735 号碱基,连上当前这条序列的 1 到 1177 号碱基,连上
AF018431 这条序列的 1 到 45 号碱基,连上 AF018432 这条序列的 658 到 732 号碱基和 884
到 954 号碱基,以及 1391 到 1447 号碱基。后面给出了基因的名字,dut。
mRNA 给出了拼凑出上面基因的各个片段中外显子的位置。也就是说,GENE 里的片段
拼在一起是完整的基因,mRNA 里的片段拼在一起就等于完成了剪切的过程,相当于成熟
mRNA。值得注意的是,剪接后形成的 mRNA 有两种,其中一种比另一种在前端多了一个
外显子。多的这一段将被翻译成定位线粒体的信号肽,从而翻译出线粒体型的蛋白质。而另
一种没有信号肽的将形成细胞核型蛋白质。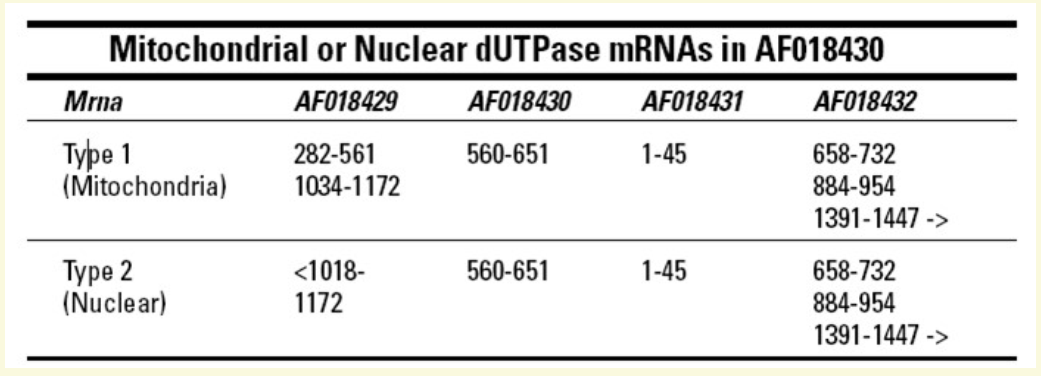
上表清晰的列出了四个片段中所有外显子的位置。能够清楚的看到,线粒体型的比细胞
核型的多了一个翻译信号肽的外显子,其他的翻译成熟肽段的外显子都是一样的。虽然信号
肽最终也会被切掉,但是由此产生了两种亚细胞定位的蛋白质。有信号肽的会到线粒体中去,
没有的将留在细胞核里。
最后 exon 条目明确的告诉我们,当前这条序列里 560 到 651 号碱基是 dut 基因的第三
个外显子。至此,大家应该看得出来,解读真核生物的 DNA 序列要比原核生物复杂得多。
但是,只要你熟知基因的结构和 Genbank 的存储方式,这本天书不难看懂。
一级核酸数据库:基因组数据库 Ensemble
在查看人的整个基因组之前,需要先搞清楚几件事。人的基因组有 33 亿个碱基分布在
23 个染色体上。我们现在已经获得了人的全基因组序列。起初拿到手的就是 33 亿个字母,
下一步面临的巨大挑战就是给它们添加注释,也就是做一个详细的 FEATURES 表。全世界
每时每刻关于人类基因及其功能都有新的发现。研究基因的方法五花八门,层出不穷,不可
能全部学会,只能是用到哪学到哪!
我们从 Ensembl 数据库(http://www.ensembl.org)查看人的基因组。Ensembl 是由欧洲
生物信息学研究所 EBI 和英国桑格研究院合作开发的。它收入了各种动物的基因组,特别
是那些离我们人类近的脊椎动物的基因组。这些基因组的注释都是通过配套开发的软件自动
添加的。Ensembl 主页左下角有人,老鼠,斑马鱼这三个点击率最高的基因组的快速链接。
其中,人的基因组有两个。右边是 2009 年获得的基因组信息,左边是 2013 年重新测序获得
的基因组信息。我们看右边这个最新的。
点击进入之后,我们点这个查看染色体,就可以看到人的所有染色体的图例。不知到大
家还记不记得,之前看到的某些信息似乎和 15 号染色体有点儿什么关系!没错,前面一直
研究的那个编码 dUPTase 的 dut 基因就在 15 号染色体上。点一下 15 号染色体,在弹出窗口
中选择染色体概要(chromosome summary)。这时我们会得到 15 号染色体的一个一览图。里
面包括编码蛋白的基因、非编码基因、假基因分别在染色体上不同区段内的含量,以及 GC
百分比(红线),和卫星 DNA 百分比(黑线)。染色体统计表给出了 15 号染色体的长度,
以及各种类型的基因的个数。
从 Genbank 我们了解到,dut 基因的第三号外显子位于 15 号染色体的长臂条带 21.1 附
近。所以我们进一步进入这个条带看一下。点击条带 21.1,选择区间链接。这时,这个区间
内所有的基因就都被显示在一张图上。如果眼力好的话,可以从这个图谱上直接找到 dut 基
因,并以他为中心放大。如果找不到,也可以通过搜索条输入基因的名字进行查找。
在以 dut 基因为中心显示的放大图谱中,点击 dut 或者对应的区域,在弹出的概况窗口
中选择 Ensemble 数据库的检索号。之后就会出现 dut 基因在 Ensemble 数据库中的详细记录。
一级核酸数据库:微生物宏基因组数据库 JCVI
微生物宏基因组数据库是非常有用的一级核酸数据库资源。说到微生物宏基因组学,不
得不介绍的是美国基因组研究所 TIGR 和克莱格反特研究所 JCVI。美国基因组研究所致力
于微生物基因组的研究,也有部分植物基因组项目。它是克莱格·凡特研究所的一部分。自
1995 年成立之初的两个基因组,至今已拥有超过 700 个基因组,而且还将更多。TIGR 是
NCBI 基因组资源的有力补充,因为它不仅拥有已完成测序的基因组,还有那些测序中的基
因组信息。在植物基因组项目中可以找到拟南芥、玉米、苜蓿和柳树的基因组信息。在微生
物与环境基因组目中,特别值得关注的是“人类微生物组计划”,HMP。
HMP 由美国 NIH 发起,由 4 个四个测序中心共同完成,其中一个就是克莱格凡特学院。
“人类微生物组计划”堪比“人类基因组计划”。我们目前认知的微生物不到 1%,生活在我
们肠道中的微生物细胞,是人体细胞的 10 倍。这些微生物基因组之和是人类基因组的 100
倍。微生物影响并超越我们的生老病死,有一天人死了,但身体中的微生物却还活着。除了
近年来少量的有关糖尿病等与肠道微生物的研究外,我们完全不清楚肠道微生物,呼吸道微
生物,还有体表微生物等在人体内做了什么,他们的喜怒哀乐与我们的生老病死有什么关系。
所以世界上诸多科学家都呼吁完成微生物组的研究计划。HMP 就是其中之一。目前,HMP
主要包括了人类鼻腔、口腔、皮肤、胃肠道和泌尿生殖道的宏基因组样本数据和分析流程。
从 JCVI 主页(http://www.jcvi.org/)的统计表中我们可以看到不同器官中有多少微生物
基因组已被测序并被注释。点击下方的统计链接。可以得到 HMP 中已研究的所有微生物基
因组。这些微生物在人体中存在的位置,测序及注释是已完成还是在进行中。已完成的基因
组后面会有三个链接:
WGS 是全基因组鸟枪法测序项目数据库记录的链接。
SRA 是高通量测序数据库记录的链接。这两个链接里记录的是测序的信息。相比之下对
大家更为有意义的是
ANNOTATION 链接里的内容,他列出了某个基因组在 Genbank 中所有注释的链接。
比如微生物 Acinetobacter radioresistens SK82 的基因组共分成 82 条序列记录在 Genbank 数据
库中。通过前面章节的学习,解读这些序列并不困难。
二级核酸数据库
二级核酸数据库包括的内容非常多。其中 NCBI 下属的三个数据库经常会用到。他们是
RefSeq 数据库,dbEST 数据库和 Gene 数据库。RefSeq 数据库,也叫参考序列数据库,是通
过自动及人工精选出的非冗余数据库,包括基因组序列、转录序列和蛋白质序列。凡是叫
ref 什么的数据库都是非冗余数据库,就是已经帮你把重复的内容去除掉了。dbEST 数据库,
也就是表达序列标签数据库,存储的是不同物种的表达序列标签。Gene 数据库以基因为记
录对象为用户提供基因序列注释和检索服务,收录了来自 5300 多个物种的 430 万条基因记
录。
此外,非编码 RNA 数据库,提供非编码 RNA 的序列和功能信息。非编码 RNA 不编码
蛋白质但在细胞中起调节作用。目前该数据库包含来源于 99 种细菌,古细菌和真核生物的
3 万多条序列。microRNA 数据库主要存放已发表的 microRNA 序列和注释。这个数据库可
以分析 microRNA 在基因组中的定位和挖掘 microRNA 序列间的关系。
关于核酸数据库就给大家介绍到这里。
=========================================
RefSeq https://www.ncbi.nlm.nih.gov/refseq/
dbEST https://www.ncbi.nlm.nih.gov/dbEST/
Gene https://www.ncbi.nlm.nih.gov/gene
ncRNA http://biobases.ibch.poznan.pl/ncRNA
microRNA http://www.mirbase.org/
=========================================
一级蛋白质序列数据库:UniProt 数据库介绍
上次课我们讲了核酸数据库。这节课我们来讲蛋白质数据库。蛋白质数据库的种类比核
酸数据库要多,但它的注释要比核酸数据库直白得多。像核酸数据库一样,蛋白质数据库也
分为一级和二级。一级蛋白质数据库又分为蛋白质序列数据库和蛋白质结构数据库。这两种
数据库里存放的都是通过实验方法直接获得的基础数据。而二级蛋白质数据库都是在一级数
据库的基础上分析加工出来的。我们首先来看一级蛋白质序列数据库。一级蛋白质序列数据
库包含三大蛋白质序列数据库,Swiss-Prot,TrEMBL 和 PIR,这三个数据库共同构成 UniProt
数据库。
Swiss-Prot 是一个人工注释的蛋白质序列数据库。它拥有注释可信度高,冗余度小的优
点。它是由欧洲生物信息学研究所 EBI 与瑞士生物信息学研究所 SIB 共同管理的。TrEMBL
也是 EBI 和 SIB 共同管理的一个数据库,他与 Swiss-Prot 的区别是:TrEMBL 里的蛋白质序
列注释是由计算机完成的,它包含了 EMBL 核酸序列数据库中为蛋白质编码的核酸序列的
所有翻译产物。换言之,TrEMBL 是通过计算机,把核酸序列数据库里能编码蛋白的核酸序
列都翻译成了蛋白质序列,然后把这些计算机翻译出来的蛋白质序列存入其中。可想而知,
这样的数据库一定是可信度低而冗余度大的。好在 TrEMBL 把已经包含在 Swiss-Prot 数据库
中的序列剔除掉了。也就是在 Swiss-Prot 里已经有人工注释的蛋白质序列在 TrEMBL 里就不
再出现了。PIR 数据库是蛋白质信息资源数据库,他设在美国 Georgetown 大学医学中心。
是一个支持基因组学,蛋白质组学和系统生物学研究的综合公共生物信息学资源。
2002 年,Swiss-Prot 和 TrEMBL 的数据库管理组与 PIR 的数据库管理组成立联合蛋白质
数据库协作组,管理联合蛋白质序列数据库,也就是 UniProt 数据库。UniProt 数据库有三个
层次。第一层叫 UniParc,收录了所有 UniProt 数据库子库中的蛋白质序列,量大,粗糙。
第二层是 UniRef,他归纳了 UniProt 几个主要数据库并且是将重复序列去除后的数据库。第
三层是 UniProtKB,他有详细注释并与其他数据库有链接,分为 UniProtKB 下的 Swiss-Prot
和 UniProtKB 下的 TrEMBL 数据库。关系稍有点复杂,但实际上我们最常用的就是 UniProtKB
下的 Swiss-Prot 数据库。
一级蛋白质序列数据库:UniProtKB 注释解读(1)
这一节我们从 UniProt 数据库查看一条蛋白质序列(http://www.uniprot.org/)。在 UniProt
数据库的首页上有一个关于 UniProtKB 数据库的统计表。可以看到,TrEMBL 数据库里存储
的序列数量远远大于 Swiss-Prot 中的。统计表里清楚的写着:TrEMBL 是自动注释的,没有
经过检查,而 Swiss-Prot 是人工注释的,并且经过检查。这是 Swiss-Prot 和 TrEMBL 最大的
区别,一定要记住。跟 NCBI 的网站一样,UniProt 数据库的首页上也有一个搜索条,选择
UniprotKB 数据库,然后输入“human dutpase”。上节课我们一直在研究 dUTPase,从 PubMed
查文献到 GenBank 查看编码这一蛋白的 dut 基因。这节课我们继续研究它。这次我们直接查
看 dUTPase 的蛋白质序列。
通过关键词搜索我们找到了很多条蛋白质序列。从蛋白质的名字来看,第一条应该是我
们想要的。Entry 这一列是蛋白质序列在 UniProtKB 数据库中的检索号,Entry_Name 是
检索名,检索号与检索名平行运行,都是一条序列在数据库中的唯一标识,两者作用相同,
只是写法不同。从检索名可以更直观的知道是哪个物种的什么蛋白质。从加星文档图标 我
们可以获知序列是被人工检查过的还是没有。也就是说,有加星文档图标的是 Swiss-Prot 中
的数据,没有的是 TrEMBL 里的。后面这几列,依次是蛋白质的名字,编码这一蛋白质的
基因的名字,所属物种以及序列长度。点击第一条序列的检索号,打开这条数据库记录。
UniProtKB 中的数据库记录分成几个部分,左侧是注释标签,点击其中某一个标签可以
直接跳转到该部分注释。上方是工具标签,可以用于和其他序列进行比较,格式转换,存储
等。工具标签下方是这条蛋白质序列的基本信息,蛋白质的名字,基因的名字,所属物种,
以及状态。这里有加星文档图标,是被人工检查过的,应该属于 Swiss-Prot 数据库。注释打
分 5 星,说明注释得很全面,并且这些注释在蛋白质水平上有实验依据。再往下就是具体的
注释内容了。
Function,功能这部分注释很详细的说明了这个蛋白质的功能。从这里可以得知
dUTPase 是一种在核酸代谢过程中的酶、它的催化反应方程式、它的辅助因子、它参与的代
谢途径等。每条注释信息都提供出处来源,让你有据可查。
Names & Taxomomy 给出了蛋白质的各种名字,包括全称、缩写以及别名。还列出了所属物种以及该物种的分类学谱系等。
Subcellular location:提供蛋白质亚细胞定位(subcellular localization)的信息。
成熟蛋白质必须在特定的细胞部位才能发挥其生物学功能。蛋白质在细胞内不同组分中的定
位即为蛋白质的亚细胞定位。亚细胞定位对蛋白质的生理功能有着直接的影响。处于合适的
亚细胞定位的蛋白质才能行使其正常的功能。目前,研究亚细胞定位的数据来源基本都是
Swiss-Prot 数据库。上节课我们从 GenBank 里查看人的 dut 基因时得知,dut 基因有两种剪切
方式,其中一种会保留前端的一段信号肽,这个信号肽会将蛋白质定位于线粒体。而没有这
段信号肽的留在了细胞核。这与 Swiss-Prot 中关于亚细胞定位的注释是一致的。我们看到,
这个蛋白有两种异构体(isofrom),一个亚细胞定位在细胞核,另一个在线粒体。
一级蛋白质序列数据库:UniProtKB 注释解读(2)
Pathology & Biotechnology:提供蛋白质突变或缺失导致的疾病及表型信息。比
如 99 位的丝氨酸会突变成丙氨酸从而导致磷酸化的缺失,相关具体研究可参考注释来源文
献。
PTM/Processing:提供蛋白质翻译后修饰或翻译后加工的相关信息。比如信号肽在
蛋白质到达指定位置之后要被剪切掉,有些氨基酸位点上会发生乙酰化、甲基化、磷酸化等
翻译后修饰。
Expression:提供了基因在 mRNA 水平上的表达信息,或者在细胞中蛋白质水平上
的表达信息,或者在不同器官组织中的表达信息。
Interaction:提供了蛋白质之间相互作用的信息。包括 UniProtKB 中直接与这个蛋
白质有两两相互作用的蛋白质序列的链接,以及这个蛋白质在各种蛋白质相互作用数据库或
蛋白质网络数据库中涉及的数据库记录链接。
Structure:提供蛋白质二级结构和三级结构信息。这里请注意,只有那些已通过实
验方法测定三级结构并且已提交到蛋白质结构数据库 PDB 的蛋白质才有结构注释。二级结
构以图形拓扑的形式呈现。三级结构列出了该蛋白质在蛋白质结构数据库 PDB 中涉及的数
据库记录链接。这些结构经常只对应蛋白质的部分序列。
Family & Domains:提供蛋白质家族及结构域信息。这个蛋白质是属于 dUTPase 家
族的。它有三个重要的区域用于和其他分子结合。此外还有与系统发生学数据库以及结构域
数据库之间的链接。
Sequence:提供蛋白质氨基酸序列信息。含有多个异构体的蛋白质会显示多条序列。
这个蛋白质有两个异构体,一个线粒体型的,一个细胞核型的。所以会显示两条序列。FASTA
按钮提供 FASTA 格式序列。
Cross-references:列出了所有通往其他含有该蛋白质信息的数据库的链接。
Publications:列出了有关这个蛋白质已发表的所有文献的信息。
Entry information:提供有关这条数据库记录的录入信息,外加一个免责声明。
Miscellaneous:杂项,包含任何无法归入前几项的内容。
Similar Proteins:在 UniRef 数据库里找到与该蛋白质在序列水平上相似的其他蛋
白质,并按相似度高低分组。如上节课所讲,凡是名字里有 ref 的数据库都是非冗余数据库,
UniRef 亦是如此,它属于 UniProt 数据库的第二个层次。
网页版的数据库记录也可以像 GenBank 一样保存成纯文本格式的本地文件。这种文本
格式的数据库记录每一行都有一个两个字母组成的条目索引,用以说明这一行记录的是什么
内容。条目索引所代表的具体内容可以参见下表
表 1. UniProtKB 纯文本(Flat File)格式数据库记录条目索引含义表
| Line code | Content | Occurrence in an entry |
|---|---|---|
| ID | Identification | Once;starts the entry |
| AC | Accession number(s) | Once or more |
| DT | Date | Three times |
| DE | Description | Once or more |
| GN | Gene name(s) | Optional |
| OS | Organism species | Once or more |
| OG | Organelle | Optional |
| OC | Organism classification | Once or more |
| OX | Taxonomy cross-reference | Once |
| OH | Organism host | Optional |
| RN | Reference number | Once or more |
| RP | Reference position | Once or more |
| RC | Reference comment(s) | Optional |
| RX | Reference cross-reference(s) | Optional |
| RG | Reference group | Once or more (Optional if RA line) |
| RA | Reference authors | Once or more (Optional if RG line) |
| RT | Reference title | Optional |
| RL | Reference location | Once or more |
| CC | Comments or notes | Optional |
| DR | Database cross-references | Optional |
| PE | Protein existence | Once |
| KW | Keywords | Optional |
| FT | Feature table data | Once or more |
| SQ | Sequence header | Once |
| blanks | Sequence data | Once or more |
| // | Termination line | Once; ends the entry |

若有收获,就点个赞吧
0 人点赞
