运行StringTie
从命令行 运行stringtie,如下所示:
stringtie <aligned_reads.bam> [options]*
该方案的主要输入是与RNA-SEQ一个BAM文件中读取,必须由他们的基因组位置(例如被分类映射的accepted_hits.bam通过生成的文件 TopHat 或输出HISAT2排序和使用samtools将其分类转换后的文件)。
运行stringtie时可以指定以下可选参数:-h /-help | 打印帮助消息并退出。 |-v | 打开详细模式,打印详细处理细节。 |-o [] <out.gtf> | 设置输出GTF文件的名称,StringTie将在其中写入汇编的脚本。可以将其指定为完整路径,在这种情况下,将根据需要创建目录。默认情况下,StringTie将GTF写入标准输出。 |-p | 指定用于脚本汇编的处理线程(CPU)的数量。预设值为1。 |-G <ref_ann.gff> | 使用参考注释文件(GTF或GFF3格式)指导组装过程。输出将包括表达的参考成绩单以及组装的任何新颖的成绩单。选项-B,-b,-e,-C(请参见下文)需要此选项。 |--rf | 假设一个搁浅的库为fr-firststrand。 |--fr | 假定一个绞合的库为fr-secondstrand。 |-l <标签> | 将设置为输出记录名称的前缀。默认值:STRG |-f <0.0-1.0> | 将预测的转录本的最小同工型丰度设置为在给定基因座处组装的最丰富的转录本的一部分。低丰度转录物通常是加工的转录物的不完全剪接的前体的产物。默认值:0.1 |-m <整数> | 设置预测成绩单所允许的最小长度。默认值:200 |-A <gene_abund.tab> | 基因丰度将以给定名称在输出文件中报告(制表符分隔格式)。 |-C <cov_refs.gtf> | StringTie输出具有给定名称的文件,其中所提供的参考文件中的所有成绩单都已被读取完全覆盖(要求-G)。 |-a | 没有拼接的结点在两端的对齐方式至少与该数量的碱基对齐,这些结点将被滤除。默认值:10 |-j | 至少应该有这么多的拼接读段在一个连接处对齐(即连接覆盖)。该数字可以是小数,因为一些读段在多个位置对齐。排列在n个位置的读段将对结覆盖率贡献1 / n。默认值:1 |-t | 此参数在组合的成绩单末尾禁用修整。默认情况下,StringTie会根据汇编成绩单覆盖率的突然下降来调整预测成绩单的开始和/或停止坐标。 |-c <浮动> | 设置预测成绩单所允许的最小阅读覆盖率。覆盖率低于此值的成绩单不会显示在输出中。默认值:2.5 |-g <整数> | 最小基因座间隙分离值。映射比该距离更近的读取将合并到同一处理包中。默认值:50(bp) |-B | 此开关启用Ballgown输入表文件(* .ctab)的输出,该文件包含-G选项给定的参考成绩单的覆盖率数据。(有关这些文件的说明,请参阅Ballgown文档。)使用此选项,StringTie可以直接替换Ballgown发行版中随附的制表程序。如果将选项-o给出为输出脚本文件的完整路径,则StringTie会将* .ctab文件写入与输出GTF相同的目录中。 |-b <路径> | 就像-B一样,此选项为Ballgown启用* .ctab文件的输出,但是这些文件将在提供的目录中创建,而不是在-o选项指定的目录中创建。注意:建议与-B / -b选项一起添加-e选项,除非StringTie GTF输出中仍需要新颖的成绩单。 |-e | 将读取比对的处理限制为仅估计和输出与-G选项给定的参考成绩单相匹配的汇编成绩单(要求-G,建议-B / -b使用)。使用此选项,将完全跳过没有参考成绩单的阅读包,例如,当给定的参考成绩单集仅限于一组靶基因时,这可能会大大提高速度。 |-M <0.0-1.0> | 设置允许在给定基因座处存在的多核苷酸位置映射的读数的最大分数。默认值:0.95 |-x <seqid_list> | 忽略指定参考序列上的所有读取比对(因此不要尝试执行转录本组装)。参数<seqid_list>可以是单个引用序列名称(例如-x chrM)或序列名称的逗号分隔列表(例如-x'chrM,chrX,chrY')。这可以加快StringTie的速度,特别是在排除线粒体基因组的情况下,即使某些特定的RNA-Seq分析可能不关心其基因,在某些情况下其基因的覆盖率也可能很高。参考序列名称区分大小写,它们必须与目标基因组的染色体/重叠群的名称完全相同,首先将RNA-Seq序列与之对准。 |--merge | 笔录合并模式。这是StringTie的特殊使用模式,与上述的程序集使用模式不同。在合并模式下,StringTie将GTF / GFF文件列表作为输入,并将这些成绩单合并/组合为一组非冗余的成绩单。在新的差异分析流水线中使用此模式来生成跨多个RNA-Seq样本的全局,统一的转录本(同种型)集。如果提供了-G选项(参考注释),则StringTie会将来自输入GTF文件的翻译片段与参考转录本组合在一起。
在merge模式下可以使用以下附加选项:-G <guide_gff> | 要包含在合并中的参考注释(GTF / GFF3) |-o <out_gtf> | 合并成绩单GTF的输出文件名(默认值:stdout) |-m <min_len> | 合并中要包括的最小输入笔录长度(默认值:50) |-c <min_cov> | 合并中包括的最小输入笔录覆盖范围(默认值:0) |-F <min_fpkm> | 合并中包含的最小输入笔录FPKM(默认值:0) |-T <min_tpm> | 合并中包含的最小输入笔录TPM(默认值:0) |-f <min_iso> | 最小同种型分数(默认值:0.01) |-i | 用保留的内含子保留合并的转录本(默认值:除非有确凿的证据,否则不保留) |-l <标签> | 输出记录的名称前缀(默认值:MSTRG) | |
输入文件
StringTie将按参考位置排序的二进制SAM(BAM)文件作为输入。该文件包含拼接的读取比对,可以直接由TopHat等程序生成,也可以通过对HISAT2的输出进行转换和排序来获取。我们建议使用HISAT2,因为它是一种快速而准确的对准程序。必须使用samtools程序对HISAT2生成的SAM格式的文本文件进行排序并将其转换为BAM格式 :
samtools view -Su alns.sam | samtools sort - alns.sorted
上述命令(alns.sorted.bam)生成的文件可用作StringTie的输入。
输入SAM文件中的每个剪接的读段比对(即,至少一个连接点的比对)都必须包含标签 XS,以指示产生产生测序序列的RNA的基因组链。由TopHat和HISAT2生成的路线(使用—dta选项运行时)已包含此标签,但是如果使用其他读取映射器,则应检查是否已包含此XS标签用于拼接路线。
注意:确保使用—dta选项运行HISAT2以进行对齐,否则结果会受到影响。
作为一种选择,可以将GTF / GFF3格式的参考注释文件提供给StringTie。在这种情况下,StringTie将更喜欢使用注释文件中的这些“已知”基因,对于表示的基因,它将计算覆盖率,TPM和FPKM值。它还将产生其他转录本,以解释注释未涵盖(或解释)的RNA-seq数据。请注意,如果选项-e不使用参考成绩单,则必须将其完全读入以包含在StringTie的输出中。在这种情况下,还将打印由StringTie从数据汇编而未出现在参考文件中的其他成绩单。
输出文件
主要产出
- Stringtie的主要输出是包含汇编成绩单的GTF文件
- 制表符分隔格式的基因丰度
- GTF格式的与参考注释完全匹配的笔录
- 输入到Ballgown所需的文件(表格),使用它们来估计差异表达
- 在合并模式下,一组GTF文件中的合并的GTF文件
1. StringTie的主要GTF输出StringTie的主要输出
是一个基因转移格式(GTF)文件,其中包含StringTie从RNA-Seq数据组装而成的转录本的详细信息。GTF是GFF(基因查找格式,也称为通用特征格式)的扩展,与GFF2和GFF3非常相似。GTF输出的9列的字段定义可以在 Ensembl网站上找到。以下是StringTie汇编的笔录示例,如GTF文件所示(在框内向右滚动以查看完整的字段内容):
seqname source feature start end score strand frame attributeschrX StringTie transcript 281394 303355 1000 + . gene_id "ERR188044.1"; transcript_id "ERR188044.1.1"; reference_id "NM_018390"; ref_gene_id "NM_018390"; ref_gene_name "PLCXD1"; cov "101.256691"; FPKM "530.078918"; TPM "705.667908";chrX StringTie exon 281394 281684 1000 + . gene_id "ERR188044.1"; transcript_id "ERR188044.1.1"; exon_number "1"; reference_id "NM_018390"; ref_gene_id "NM_018390"; ref_gene_name "PLCXD1"; cov "116.270836";...
每列值的说明:
- seqname:表示此转录本的染色体,重叠群或支架。这里的转录本在X染色体上。
- source:GTF文件的源。由于此示例是由StringTie制作的,因此此列仅显示了“ StringTie”。
- feature:功能类型;例如外显子,转录本,mRNA,5’UTR)。
- start:使用基于1的索引开始特征(外显子,转录本等)的起始位置。
- end:要素的结束位置,使用基于1的索引。
- score:成绩单的置信度得分。当前不使用此字段,并且如果笔录与读取的比对束连接,则StringTie报告恒定值1000。
- strand:如果转录本位于正链上,则为“ +”。如果转录本位于反向链上,则为“-”。
- frame:CDS功能的框架或相位。StringTie不使用此字段,而仅记录一个“。”。
- attribute:标签值对的分号分隔列表,提供有关每个功能的附加信息。根据实例是转录本还是外显子,以及转录本是否与用户提供的参考注释文件相匹配,属性字段的内容将有所不同。以下列表描述了此列中显示的可能属性:
- gene_id:基于比对文件名的单个基因及其子转录本和外显子的唯一标识符。
- transcript_id:基于比对文件名的单个脚本及其子外显子的唯一标识符。
- exon_number:给定笔录中单个外显子的唯一标识符,从1开始。
- reference_id:实例匹配的参考注释中的transcript_id(可选)。
- ref_gene_id:实例匹配的参考注释中的gene_id(可选)。
- ref_gene_name:参考注释中的gene_name(可选),与实例匹配。
- cov:笔录或外显子的平均每碱基覆盖率。
- FPKM:每百万个读对中每千个转录本片段的片段。这是与该功能对齐的读取对数,通过测序的片段总数(以百万为单位)和转录本的长度(以千碱基为单位)进行标准化。
- TPM:每百万笔成绩单。这是来自该特定基因的转录本数量,首先通过基因长度,然后通过样品中的测序深度(以百万为单位)进行标准化。可在此处找到TPM和FPKM的详细说明和比较,B。Li 和C. Dewey在此处定义了 TPM 。
2.制表符分隔格式的基因丰度
如果使用该-A <gene_abund.tab>选项运行StringTie ,它将返回一个包含基因丰度的文件。制表符分隔的基因丰度输出文件有9个字段;这是StringTie使用参考注释生成的基因丰度文件的示例:
Gene ID Gene Name Reference Strand Start End Coverage FPKM TPMNM_000451 SHOX chrX + 624344 646823 0.000000 0.000000 0.000000NM_006883 SHOX chrX + 624344 659411 0.000000 0.000000 0.000000...
* 第1列/ Gene ID:基因标识符来自`-G`选件随附的参考注释。如果未提供参考,则此字段将替换为输出记录的名称前缀(-l)。* 第2列/ Gene Name:此字段包含该`-G`选项随附的参考注释中的基因名称。如果未提供参考,则此字段将填充“-”。* 第3列/ Reference:用于比对读段的参考序列的名称。等效于.SAM对齐方式中的第三列。* 第4列/ Strand:“ +”表示基因在正链上,“-”表示反向链。* 第5列/ **开始**:基因的开始位置(从1开始的索引)。* 第6列/ **结束**:基因的结束位置(从1开始的索引)。* 第7栏/ **覆盖率**:基因的每碱基覆盖率。* 第8列/ **FPKM**:以FPKM为单位的标准化表达水平(请参阅上一节)。* 第9列/ **TPM**:以RPM为单位的标准化表达水平(请参见上一节)。
3.与参考注释笔录完全匹配的笔录(GTF格式)
如果使用-C <cov_refs.gtf>选项(requires -G <reference_annotation>)运行StringTie ,它将返回一个文件,其中包含参考注释中的所有笔录,这些笔录被读取端到端完全覆盖。如上所述,输出格式是GTF文件。GTF的每一行对应于参考注释中的基因或转录本。
4.Ballgown 输入表文件
如果使用该-B选项运行StringTie ,它将返回Ballgown输入表文件,其中包含所有笔录的覆盖率数据。输出表文件与GTF主输出位于同一目录中。这些表具有以下特定名称:(1)e2t.ctab,(2)e_data.ctab,(3)i2t.ctab,(4)i_data.ctab和(5)t_data.ctab。可以在Ballgown网站的此链接上找到对Ballgown的这五个必需输入中的每一个的详细描述。
5.Merge mode: Merged GTF
如果使用该--merge选项运行StringTie ,它将输入一个GTF / GFF文件列表并将这些笔录合并/组合为一组非冗余笔录。该步骤为所有样品创建统一的转录本集,以促进下游计算不同实验条件下所有转录本的差异表达水平。输出是具有所有合并的基因模型的合并的GTF文件,但是在覆盖率,FPKM和TPM方面没有任何数字结果。然后,使用此合并的GTF,StringTie可以通过使用-e原始对齐文件集上的选项再次运行它来重新估计丰度,如下图所示。_
评估笔录集
获取有关由StringTie(基因和转录物计数的摘要,与新颖已知等)组装在转录物的详细信息,或者甚至在执行跨多个RNA测序实验组装亚型的基本跟踪的一个简单的方法,是使用gffcompare程序。该程序的基本用法信息和下载选项可在GFF实用程序页面上找到。
差异表达分析
与HISAT和Ballgown一起,StringTie可用于估计多个RNA-Seq样品之间的差异表达,并生成我们的方案文件中所述的图和差异表达表。 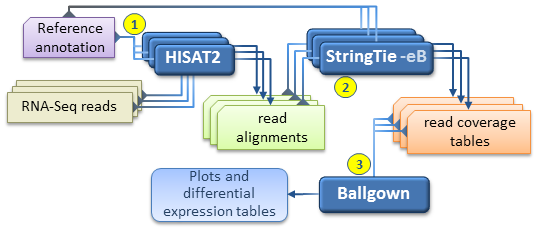
我们建议的工作流程包括以下步骤:
- 对于每个RNA-Seq样品, 使用—dta选项将读取的结果映射到具有HISAT2的基因组。强烈推荐使用映射读取当参考注释信息,它可以是嵌入在基因组中的索引(与内置—ss和—exon选项,请参阅 HISAT2手册),或在运行时单独地提供(使用在—known-剪接位点,INFILE的选项 HISAT2)。如上所述,必须对每个HISAT2运行的SAM输出进行排序,并使用samtools将其转换为BAM 。
- 对于每个RNA-Seq样品,运行StringTie组装上一步中获得的读取比对;如果参考注释可用,建议使用-G选项运行StringTie。
- 使用—merge运行StringTie,以生成在先前组装的所有 RNA-Seq样本中观察到的非冗余转录本 集合。所述 stringtie —merge模式作为输入预先针对每个样品获得的所有组装转录的文件的列表(在GTF格式),以及作为参考注释文件(-G如果可用选项)。
- 对于每个RNA-Seq样本,请使用-B / -b和-e选项运行StringTie ,以估计转录本丰度并生成Ballgown的读取覆盖率表。的-e选项不是必需的,但建议在此运行中使用此选项,以便对输入的笔录产生更准确的丰度估计。在此步骤中运行的每个StringTie都将输入在步骤1中为相应样本获得的已排序阅读比对(BAM文件)以及 -G选项以及在步骤3中由stringtie —merge生成的合并成绩单(GTF文件)作为输入。请注意,这是唯一未将-G选项与参考注释一起使用,而是与在所有样本中观察到的全局,合并的成绩单集一起使用的情况。(此步骤等效于原始Ballgown管道中描述的Tablemaker步骤。)
- 现在,可以使用Ballgown加载上一步中生成的coverage表,并对差异表达式执行各种统计分析,生成图等。
如果对新型同工型(例如样品中存在但参考注释中缺少的组装转录本)没有兴趣,或者如果目标基因只靶向了一组众所周知的感兴趣的转录本,则可以采用另一种更快的差异表达分析工作流程。分析。这个简化的方案仅需3个步骤(如下所示),因为它绕过了每个RNA-Seq样品的单独组装和 “转录本合并”步骤。
这种简化的工作流程尝试直接估算和分析参考注释文件中给定的一组已知成绩单的表达。
将StringTie与DESeq2和edgeR一起使用
DESeq2和edgeR是用于分析差异表达的两种流行的Bioconductor软件包,它们将映射到特定基因组特征(例如基因)的读取计数矩阵作为输入。我们提供了一个Python脚本(prepDE.py),可直接从StringTie生成的文件(使用-e参数运行)中提取此读取计数信息。
提供输入的方法有两种:一种选择是提供一个目录,该目录具有与StringTie协议文件中创建的目录相同的结构,以准备Ballgown。缺省情况下(无-i选项),它位于工作目录中,并命名为ballgown(请参见下文)。
./ballgown/sample1/sample1.gtf./ballgown/sample2/sample2.gtf./ballgown/sample3/sample3.gtf
或者,可以提供一个列出样本ID及其各自路径的文本文件(sample_lst.txt)。
用法:
prepDE.py [options]
生成包含用于基因和转录物的计数矩阵2个的CSV文件,使用在stringtie -e的输出中发现的覆盖值
选项:
-h,--help | 显示此帮助消息并退出 |-i INPUT,--input = INPUT,--in = INPUT | 示例子目录的父目录或.txt文件,以制表符分隔格式列出示例ID和GTF文件的路径[默认值:ballgown] |-g | 输出基因计数矩阵的位置[默认值:gener_count_matrix.csv] |-t | 输出笔录计数矩阵的位置[默认:transcript_count_matrix.csv] |-l LENGTH,--length = LENGTH | 平均读取长度[默认值:75] |-p PATTERN,--pattern =模式 | 选择样本子目录的正则表达式 |-c,-cluster | 是否将与不同基因ID重叠的基因聚类,而忽略具有geneID模式的基因(请参见下文) |-s STRING,-string = STRING | 如果StringTie分配的GeneID使用了不同的前缀[默认值:MSTRG] |-k KEY,--key = KEY | 如果是集群,此脚本分配的geneID使用什么前缀[默认:prepG] |--legend =传奇 | 如果聚类,则将图例文件映射记录输出到分配的GeneID的位置[默认值:legend.csv] |
然后可以将这些计数矩阵(CSV文件)导入R以供DESeq2和edgeR使用(分别使用DESeqDataSetFromMatrix和DGEList函数)。
协议:将StringTie与DESeq2一起使用
给定GTF列表,这些列表在合并时会重新估算,用户可以按照以下协议使用DESeq2进行差异表达分析。要从原始读取生成GTF,请遵循StringTie协议文件(直到Ballgown步骤)。
如上所述,prepDE.py或者接受一个.txt(sample_lst.txt列出样品ID和基转移酶路径)文件或默认期望一个长礼服通过StringTie与运行产生的目录-B选项。以下步骤引导我们通过生成基因和转录本的计数矩阵,将这些数据导入DESeq2,并进行一些基本分析。
- 使用prepDE.py生成计数矩阵
- 假定由StringTie $ python生成的默认Ballgown目录结构
python prepDE.py - GTF样本ID和路径列表(sample_lst.txt)
$ python prepDE.py -i sample_lst.txt
- 假定由StringTie $ python生成的默认Ballgown目录结构
安装R和DESeq2。安装R后,请在R上安装DESeq2:
`> source(“https://bioconductor.org/biocLite.R“)biocLite(“DESeq2”)`
在R中导入DESeq2库
> library("DESeq2")加载基因(/转录物)计数矩阵和标签 注意:PHENO_DATA文件包含每个样本的信息,例如性别或人口。导入此文件的确切方法取决于文件的格式。
`> countData <- as.matrix(read.csv(“gene_count_matrix.csv”, row.names=”gene_id”))colData <- read.csv(PHENO_DATA, sep=”\t”, row.names=1)`
检查colData中的所有样品ID是否也在CountData中并匹配其顺序
> all(rownames(colData) %in% colnames(countData))[1]是
`> countData <- countData[, rownames(colData)]all(rownames(colData) == colnames(countData)) `[1]是
从计数矩阵和标签创建DESeqDataSet
> dds <- DESeqDataSetFromMatrix(countData = countData, colData = colData, design = ~ CHOOSE_FEATURE)对DESeq2运行默认分析并生成结果表
`> dds <- DESeq(dds)res <- results(dds)`
按调整后的p值排序并显示
> (resOrdered <- res[order(res$padj), ])
_
组装超级读物
尽管StringTie主要是由基因组指导的方法,但它可以借鉴从头基因组组装获得的算法技术,以帮助进行转录本组装。它的输入不仅可以包括基于引用的汇编程序使用的拼接阅读比对,还可以包含更长的重叠序列,即它是从转录本的明确,无分支的部分重新组装而成。如果您的RNA-Seq数据已配对,则可以使用我们的方法从其末端序列(我们称为超读)重建RNA-Seq片段。这是为StringTie的输入准备对齐方式的可选步骤,但它可以提高转录组组装的准确性。要使用此步骤,您首先需要安装MaSuRCA基因组组装器。MaSuRCA的超级读取模块可以双向扩展每个读取,只要此扩展是唯一的即可。我们在此处提供的superreads.pl脚本可识别属于同一超级读取的成对读取,并提取包含该对的序列及其之间的序列;即原始DNA片段的完整序列。因此,例如,如果原始RNA-seq数据包含来自300-bp片段文库的成对的100-bp读段,则这些步骤会将其中许多对转换为单个300-bp的超读段。 下面介绍了superreads.pl脚本的用法。
用法:superreads.pl
参数:superreads.pl脚本
的前两个参数是fastq格式的文件,分别包含每个片段中第一次读取和第二次读取的序列。它们可以是纯文本的fastq文件,也可以是压缩的文件(带有gzip或bzip2),第三个参数表示系统上MaSuRCA软件包的安装目录。选项:
| -t <线程数> | 设置要使用的线程数。默认值:10 |
| -j
| -s <步骤> | 随着进度的进行,superreads.pl脚本将打印成功完成的步骤。如果由于某种原因停止了组装过程,则无需重做所有成功完成的步骤,并且可以在脚本未完成的第一步重新启动。默认值:1。 |
| -r <成对读取前缀> | 根据MaSuRCA的要求,为配对的读数设置前缀。默认值:pe。 |
| -f <片段大小> | 指定平均库插入长度。默认值:300。 |
| -d <标准偏差> | 指定库插入长度的标准偏差。如果标准偏差未知,则将其设置为平均值的15%左右。默认值:45 |
| -l <超级读取文件名> | 指定组装的超级读取文件的名称。默认值:LongReads.fq。 |
| -u
然后可以使用您的偏好的读取映射器 将superreads.pl脚本的输出文件(组合的超级读取文件,以及两个包含未组合的成对读取的文件)与参考基因组对齐。例如,您可以将它们与TopHat2对齐,如下所示:
tophat [选项] *
提供更好的翻译建议

若有收获,就点个赞吧
0 人点赞
