单细胞RNA测序(scRNA-Seq)技术的迅速扩散刺激了检测转录一致群体的多种计算方法的发展。尽管检测异质性的算法的复杂性有所增加,但是大多数算法都需要大量的用户调整,严重依赖于降维技术,并且无法扩展至超大型数据集。辛辛那提大学的研究人员先前描述了一种多步骤算法,即迭代聚类和引导基因选择(ICGS),该算法应用基因内相关和杂交聚类从直观的图形用户界面中唯一解析新颖的转录相干细胞群体。
在这里,他们描述了ICGS的新版本,当应用于完善的基准测试时,其性能优于最新的scRNA-Seq检测工作流程。这种方法结合了多种互补亚型检测方法(HOPACH,稀疏NMF,簇“适应性”,SVM)来解析稀有和常见细胞状态,同时最大程度地减少了由于供体或批次效应引起的差异。利用来自多个细胞图谱的数据,研究人员表明,PageRank算法可有效地对超大型scRNA-Seq数据集进行下采样,而不会丢失极为稀有或转录相似但又不同的细胞类型,同时还能恢复新颖的转录不同细胞群体。他们认为,这种新方法在可再现地解决复杂数据集中的隐藏细胞群体方面具有巨大的前景。
ICGS2针对多种可选无监督scRNA-Seq算法的性能
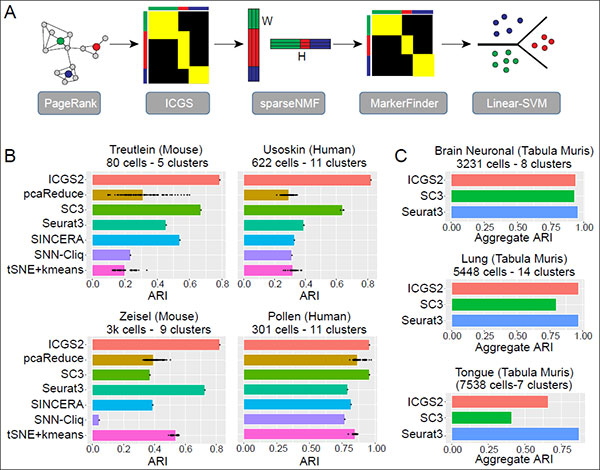
A)用于单细胞RNA-Seq群体预测的ICGS2工作流程概述。这些步骤包括:1)PageRank-Down-sample(可选),2)特征选择(ICGS),3)降维(Sparse-NMF),4)群集细化/排除(“ fitness”)和5)群集分配(线性SVM)。B)将ICGS2与先前评估过的算法进行比较,并比较大小和复杂度不同的基准数据集,以检测先前定义的细胞群。通过将作者注释的单元格到集群的分配与使用调整后的兰德指数(ARI)的每种算法获得的分配进行比较,来评估每种方法的性能。C) 将ICGS2与图B中针对烟粉虱组织scRNA-Seq(SMARTSeq2)的性能最高的方法进行比较,使用汇总ARI来说明合成的子类(相应的ARI值请参见图S1B,表中的簇数请参见表S1) )。
可用性 – ICGS2是用Python实现的。源代码和文档可在以下位置获得:http : //altanalyze.org
Venkatasubramanian M,Chetal K,Schnell D,Atluri G,Salomonis N.(2020)通过顺序杂交聚类和NMF解决了成千上万个细胞的单细胞异质性。生物信息学 [Epub提前出版]。[ 摘要 ]

若有收获,就点个赞吧
0 人点赞
