高维单细胞RNA测序数据分析工具
发布者: RNA-Seq的博客 中 评论刊物 3天前 486次查看
在单细胞水平上分析转录组的高通量技术的发展突破,已帮助生物学家了解细胞群体,疾病状态和发育谱系的异质性。但是,这些单细胞RNA测序(scRNA-seq)技术会产生大量数据,这给分析和解释带来了挑战。另外,由于不完全的RNA捕获,PCR扩增偏差和/或特定于患者或样品的批次效应,scRNA-seq数据集通常包含噪声的技术来源。如果不解决,这种技术噪音可能会使数据的分析和解释产生偏差。为了应对这些挑战,已经开发了一套计算工具来处理,分析和可视化scRNA-seq数据集。尽管任何给定的scRNA-seq分析的具体步骤可能会因所询问的生物学问题而有所不同,但大多数分析都使用核心工作流程。通常,将原始测序读数处理到基因表达矩阵中,然后对其进行标准化和缩放以消除技术噪音。接下来,根据基因表达模式的相似性将细胞分组,可以将它们概括为二维或三维图,以便在散点图中可视化。然后可以进一步分析这些数据,以深入了解目标样品中的细胞类型或发育轨迹。原始测序读数被处理成基因表达矩阵,然后对其进行标准化和缩放以消除技术噪音。接下来,根据基因表达模式的相似性将细胞分组,可以将它们概括为二维或三维图,以便在散点图中可视化。然后可以进一步分析这些数据,以深入了解目标样品中的细胞类型或发育轨迹。原始测序读数被处理成基因表达矩阵,然后对其进行标准化和缩放以消除技术噪音。接下来,根据基因表达模式的相似性将细胞分组,可以将它们概括为二维或三维图,以便在散点图中可视化。然后可以进一步分析这些数据,以深入了解目标样品中的细胞类型或发育轨迹。
Breakthroughs in the development of high-throughput technologies for profiling transcriptomes at the single-cell level have helped biologists to understand the heterogeneity of cell populations, disease states and developmental lineages. However, these single-cell RNA sequencing (scRNA-seq) technologies generate an extraordinary amount of data, which creates analysis and interpretation challenges. Additionally, scRNA-seq datasets often contain technical sources of noise owing to incomplete RNA capture, PCR amplification biases and/or batch effects specific to the patient or sample. If not addressed, this technical noise can bias the analysis and interpretation of the data. In response to these challenges, a suite of computational tools has been developed to process, analyse and visualize scRNA-seq datasets. Although the specific steps of any given scRNA-seq analysis might differ depending on the biological questions being asked, a core workflow is used in most analyses. Typically, raw sequencing reads are processed into a gene expression matrix that is then normalized and scaled to remove technical noise. Next, cells are grouped according to similarities in their patterns of gene expression, which can be summarized in two or three dimensions for visualization on a scatterplot. These data can then be further analysed to provide an in-depth view of the cell types or developmental trajectories in the sample of interest.
具有离散像元类型的数据集中的像元聚类
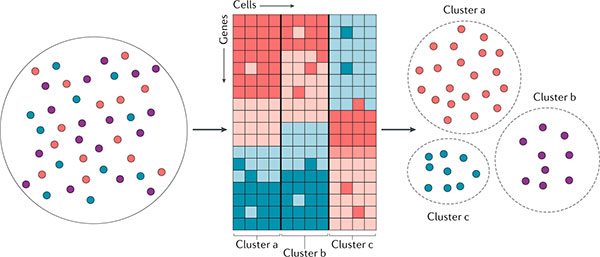
单细胞RNA测序分析的一个重要目标是通过识别存在的不同亚群来解决数据集的细胞异质性。如热图所示,细胞聚类根据基因表达模式的相似性,将来自异类数据集的细胞识别并分组为簇。这些细胞簇通常对应于数据集中存在的不同细胞类型。
An important objective of single-cell RNA sequencing analysis is to resolve the cellular heterogeneity of a dataset by identifying the different subpopulations present. Cell clustering identifies and groups cells from a heterogeneous dataset into clusters, according to similarities in their patterns of gene expression, as illustrated in the heatmap. These cell clusters usually correspond to different cell types present in a dataset.
Wu Y,Zhang K.(2020)用于分析高维单细胞RNA测序数据的工具。Nat Rev Nephrol [Epub提前发行]。[ 摘要 ]

若有收获,就点个赞吧
0 人点赞
