in Review Publications 8 days ago 722 Views
Single-cell RNA sequencing (scRNA-seq) identifies cell subpopulations within tissue but does not capture their spatial distribution nor reveal local networks of intercellular communication acting in situ. A suite of recently developed techniques that localize RNA within tissue, including multiplexed in situ hybridization and in situ sequencing (here defined as high-plex RNA imaging) and spatial barcoding, can help address this issue. However, no method currently provides as complete a scope of the transcriptome as does scRNA-seq, underscoring the need for approaches to integrate single-cell and spatial data. Researchers from Stanford University discuss efforts to integrate scRNA-seq with spatial transcriptomics, including emerging integrative computational methods, and propose ways to effectively combine current methodologies.
Adding spatial information to transcriptomes: integration of
single-cell and spatial transcriptomics data
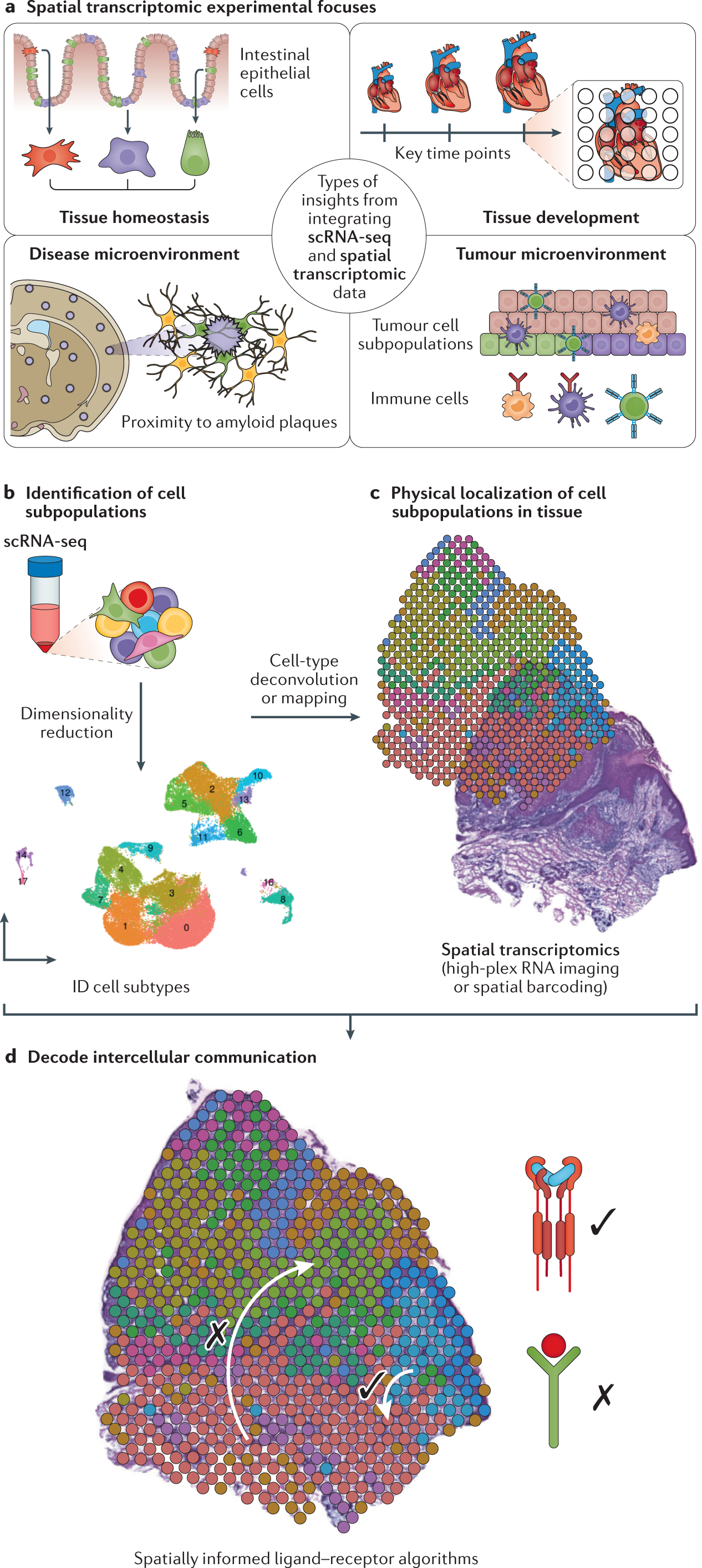
a | ‘Tissue homeostasis’ refers to elucidating spatial division of discrete cellular subtypes in a healthy tissue at a singular time point, for example, in the intestinal epithelium. ‘Tissue development’ refers to the study of how the spatial transcriptome changes in tissue at key stages in the development of a tissue. ‘Disease microenvironment’ refers to elucidating the spatial transcriptome in diseased and injured tissue niches with an eye towards proximity to relevant biological features, for example, proximity to amyloid plaques in brain tissue of patients with Alzheimer disease. ‘Tumour microenvironment’ refers to the study of spatial architecture of tumours and their interface with other cell subtypes in their environment. b | Workflows combining single-cell RNA sequencing (scRNA-seq) and spatial transcriptomic techniques begin by establishing cell subtypes typically through dimensionality reduction and clustering of scRNA-seq data. c | Deconvolution and mapping are used to localize cell subpopulations. Deconvolution is typically applied to spatial barcoding data, and mapping is typically applied to single-cell resolution spatial data (that is, high-plex RNA imaging (HPRI) data) to localize scRNA-seq subpopulations. d | Algorithms that evaluate spatial arrangement of localized subpopulations can further assess ligand–receptor interactions predicted from scRNA-seq data.
Longo SK, Guo MG, Ji AL, Khavari PA. (2021) Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nat Rev Genet [Epub ahead of print]. [article]
https://www.rna-seqblog.com/integrating-single-cell-and-spatial-transcriptomics-to-elucidate-intercellular-tissue-dynamics/

若有收获,就点个赞吧
0 人点赞
