蛋白质的结构总览
1)氨基酸序列
2)周期性的结构构象
3)整条多肽链的三维空间结构
4)多个亚基形成的复合体结构
蛋白质的二级结构
二级结构为蛋白质经过折叠,构成的规则的片段。
其中包括:
1)螺旋,最常见的就是alpha 螺旋,此外还有其他一些类型的折叠;
2)beta 折叠,beta 折叠由beta 折片(beta-strand)平行排列构成;
3)无规卷曲coil:无规律松散结构。
4)beta 转角turn:如果肽链发生了急转弯(角度大于90度),这个转弯结构叫做beta 转角。
蛋白质的二级结构经常用图形来进行描述。
可以在PDB 数据库中进行查看。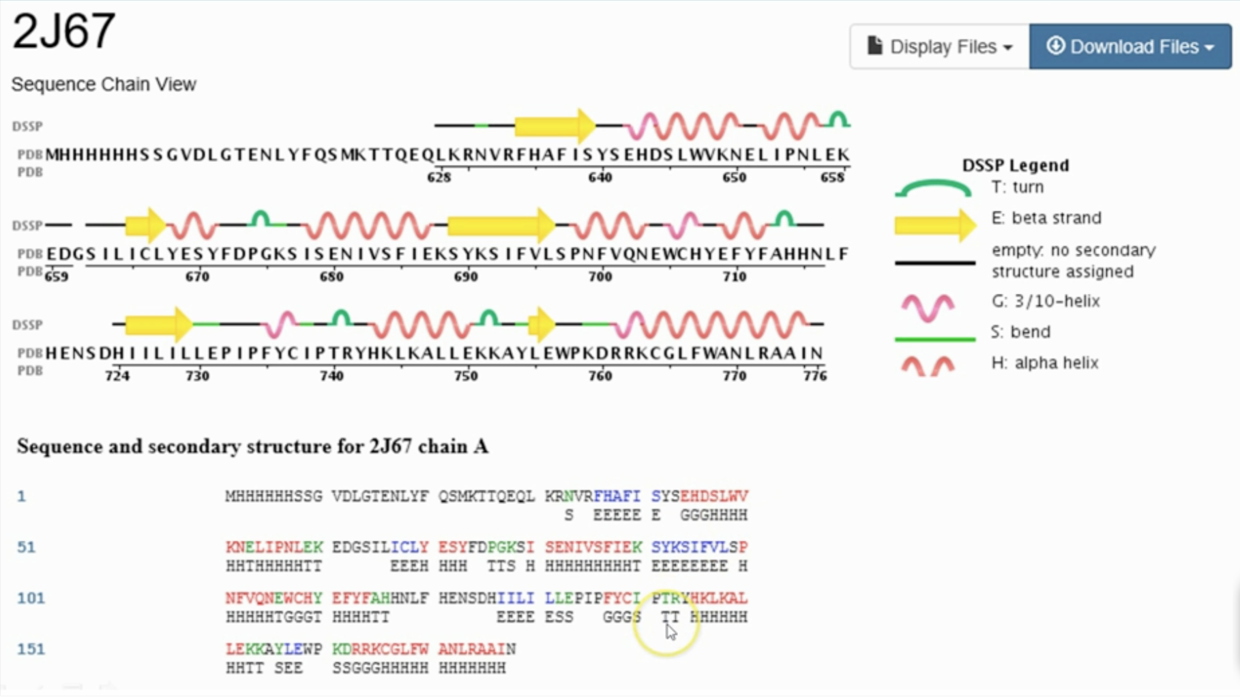
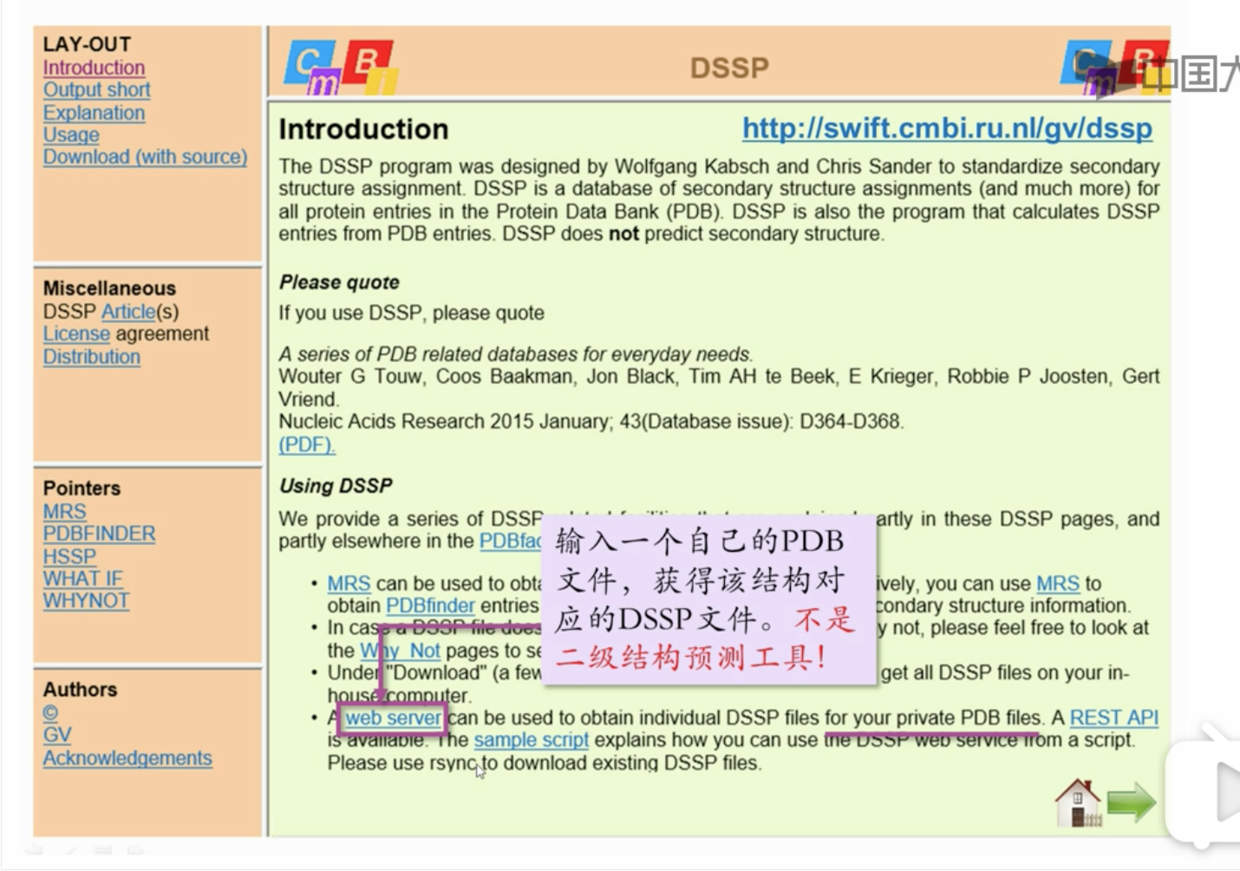
具体的二级结构信息,是通过DSSP,definition of secondary structure of proteins,即蛋白质二级结构定义词典,通过对二级结构的定义从已经测定的三级结构的蛋白质各个位置指认为何种二级结构。
- DSSP 文件
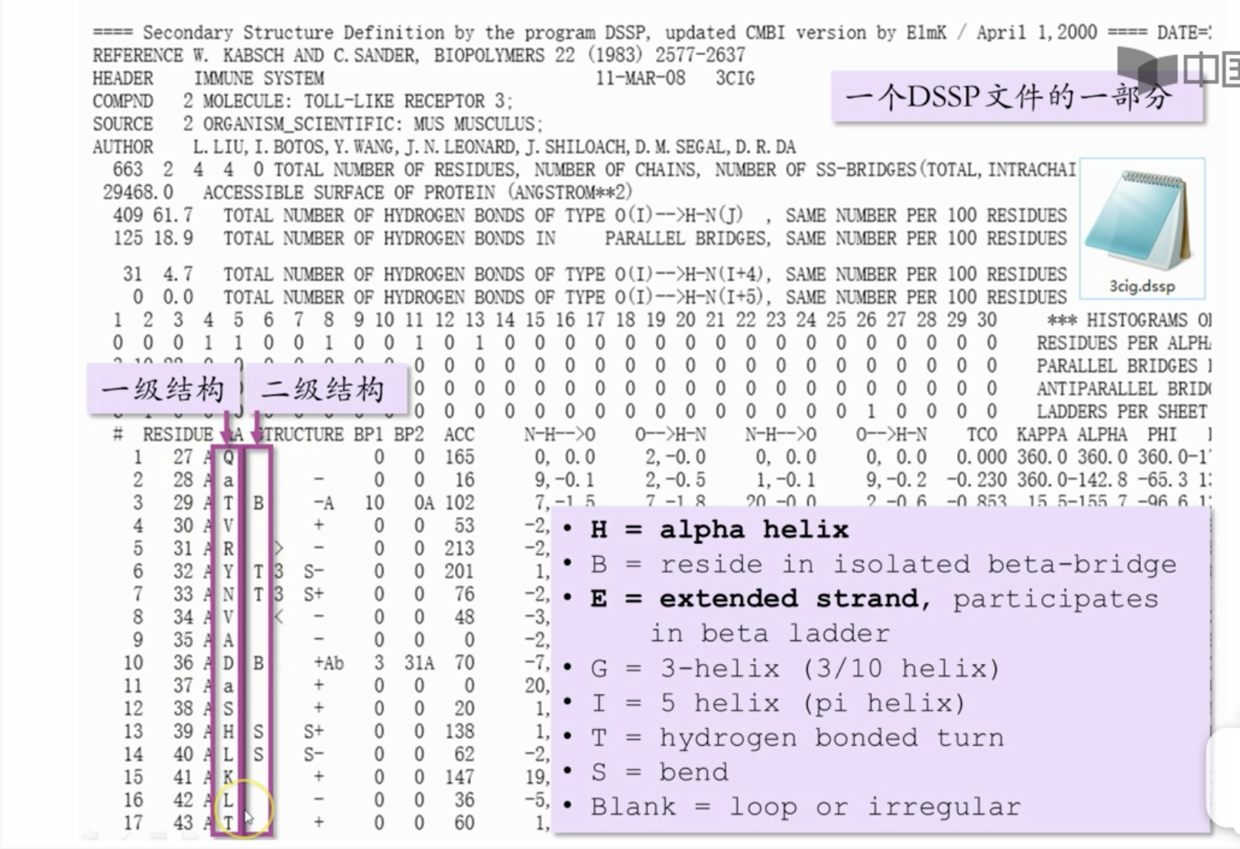
H 为alpha 螺旋,E 为beta 折片。 - 也可以在DSSP 上创建PDB 对应的DSSP 文件
- 下载蛋白质二级结构的fasta 文件

山大的biotools 工具仅限于内网使用。(通过pdb 编号,获取其fasta 文件信息)但或许互联网上有其他应用,或者自己也可以用perl 写一个。
- 预测蛋白质二级结构

PSIPRED 平台
http://bioinf.cs.ucl.ac.uk/psipred/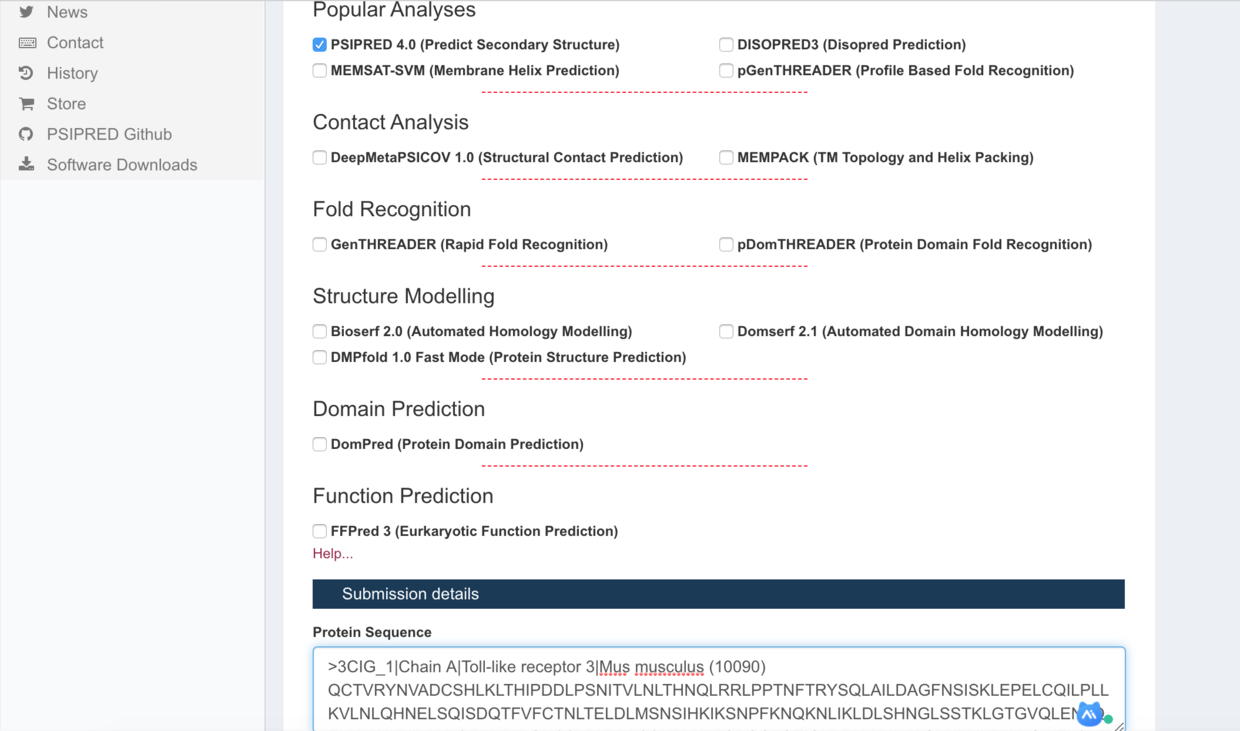
一般来说预测耗时很长,可以给个邮箱地址(需要使用工作或学校邮箱),结果会发送到邮箱里。
- 我们可以使用一条已知二级结构的氨基酸序列进行测试
其中,粉红色为alpha 螺旋,黄色为beta 折片。(目前预测软件也大部分只能预测这两种)
分别为,可信度、图形描述、二级结构、一级结构
另外也可以下载文本格式的结果文件
- psipred 与pdb 真实结果对比
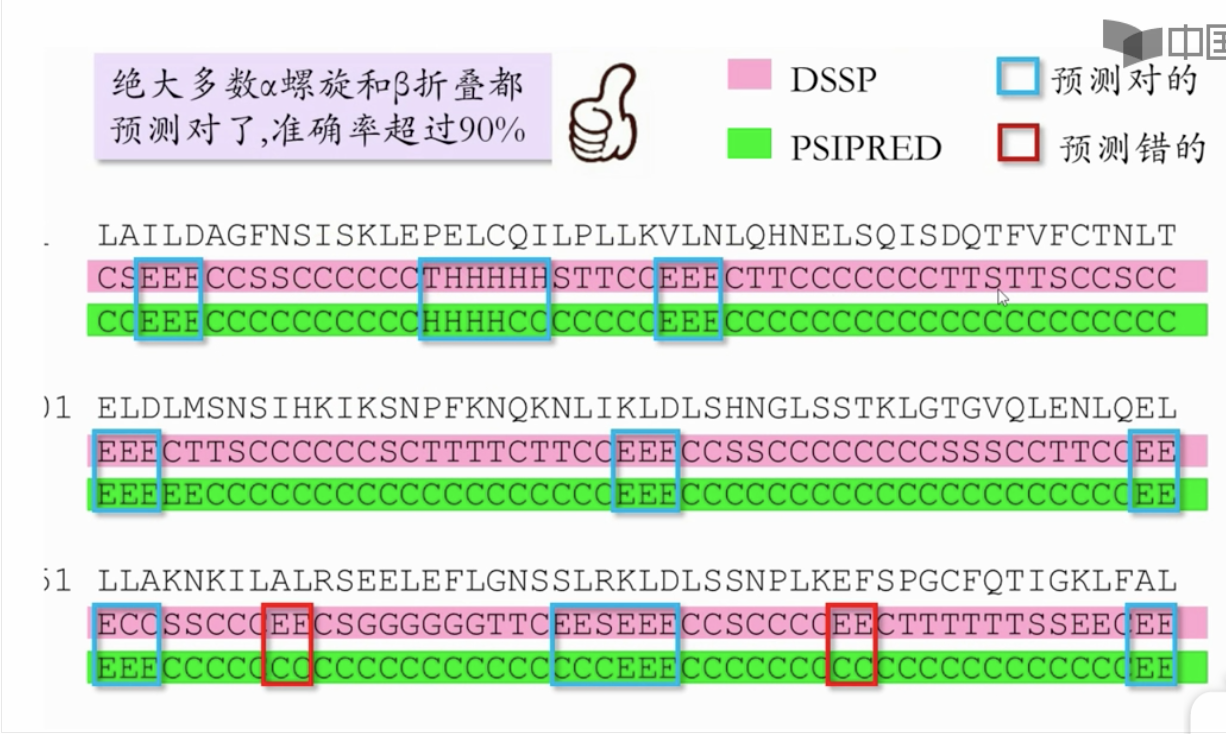
对于没有二级结构的氨基酸序列,可以多使用几个二级结构预测软件,综合所以软件结果,选择最可靠的结果。
蛋白质的三级结构
三级结构指的是蛋白质的整条多肽链的三维空间结构。
既可以通过PDB ID 或作者等信息直接搜索,也可以采用精确搜索的方式。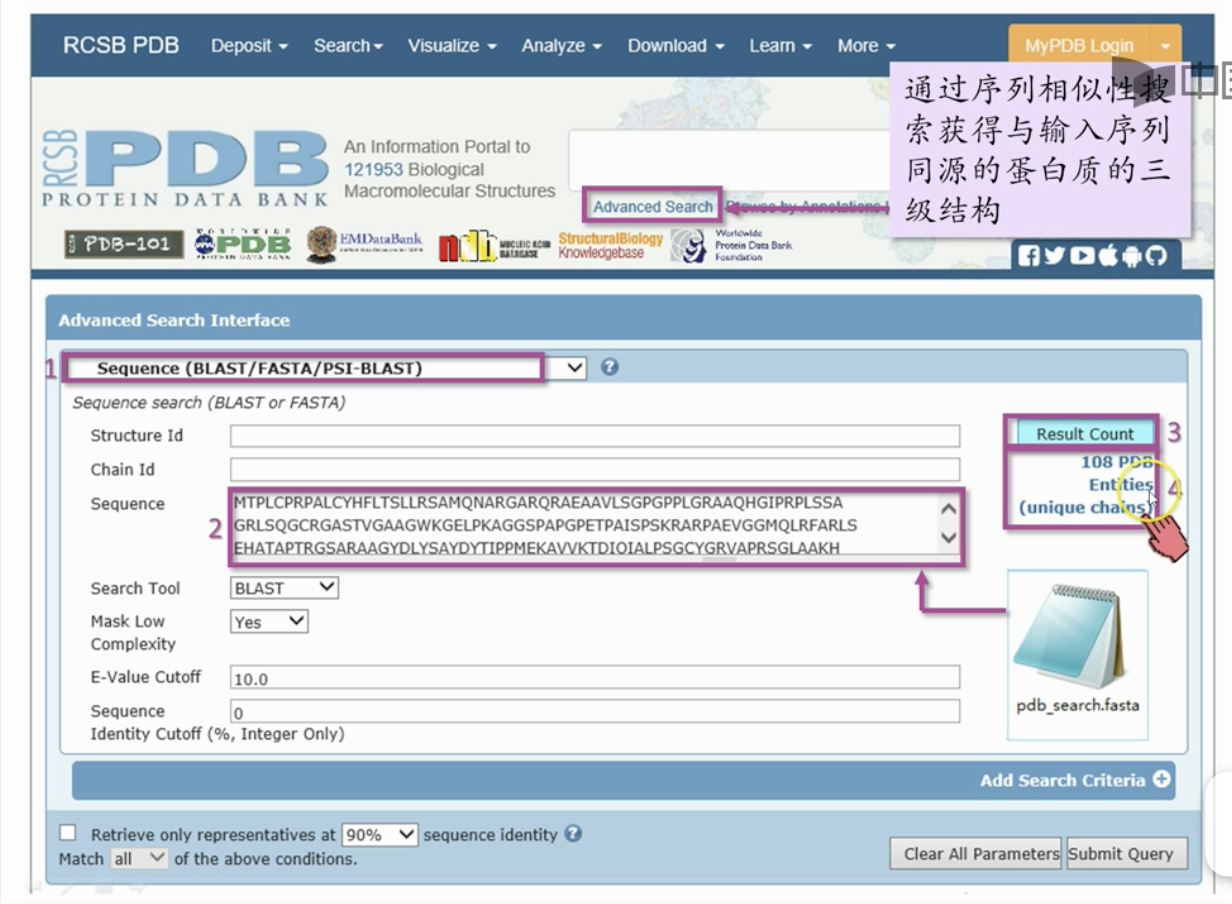
搜索结果。“未解析”的原因可能是受限于实验技术,这在pdb 中是非常常见的。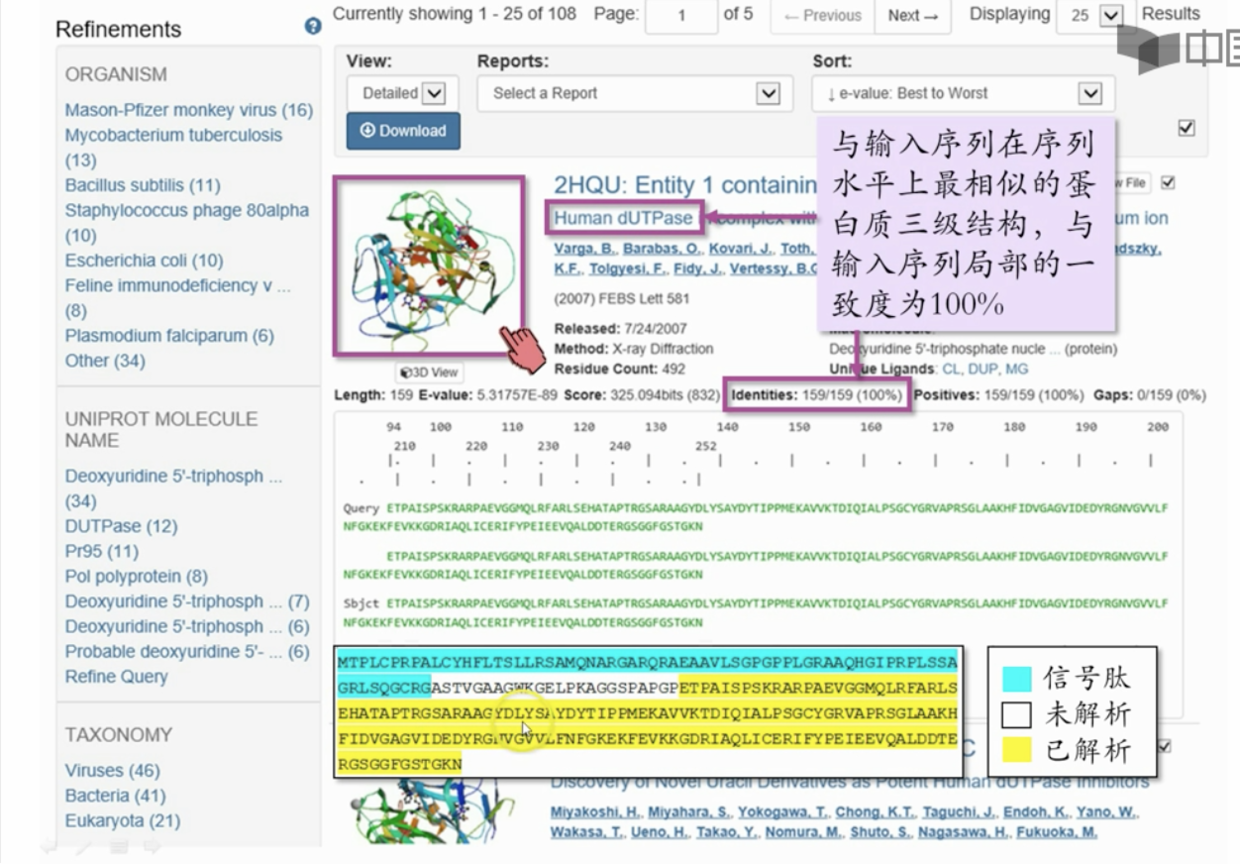
解读PDB 文件信息。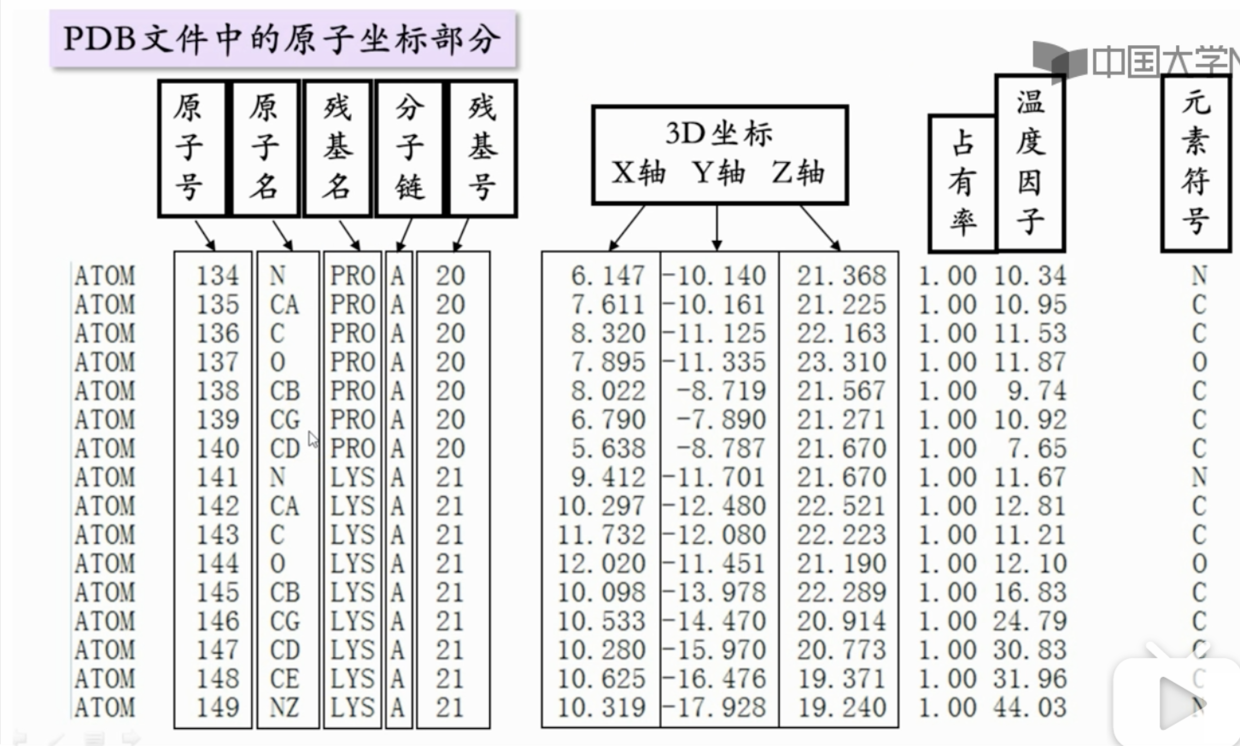
PDB 文件可以导入到蛋白质3D 可视化软件以进一步操作,如VMD、Pymol、Maestro 等等。
三级结构可视化软件VMD
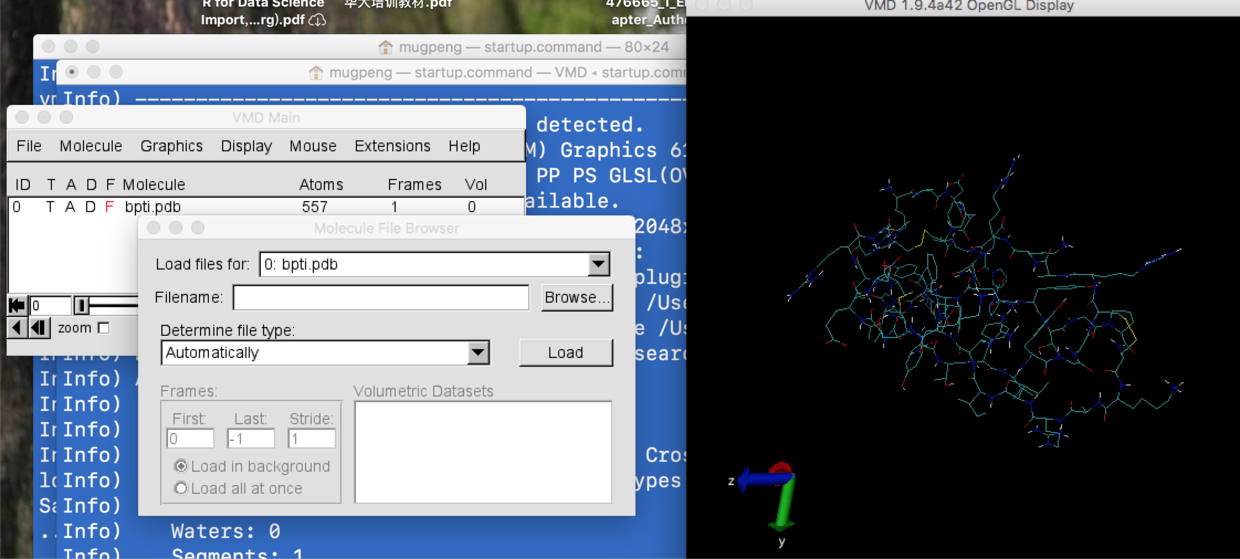
鼠标的用法
其中,可以通过左键拖拽任意划动。通过右键在平面内翻转。通过滚轮,控制显示大小。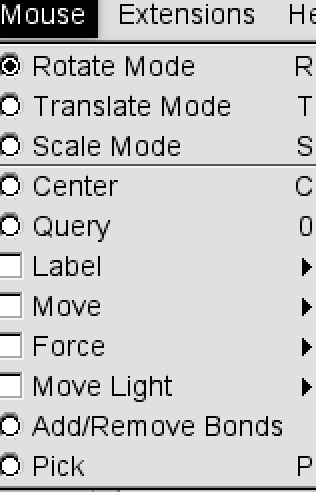
还可以调整鼠标的模式用法。
通常有以下几个模式。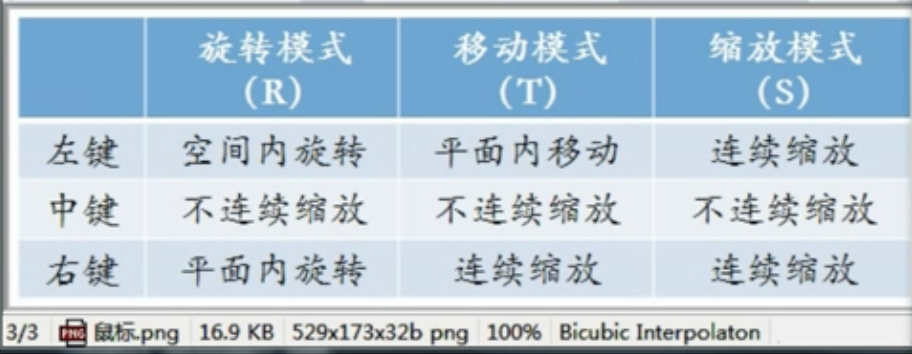
除此之外,还有中心模式等等。
显示方式
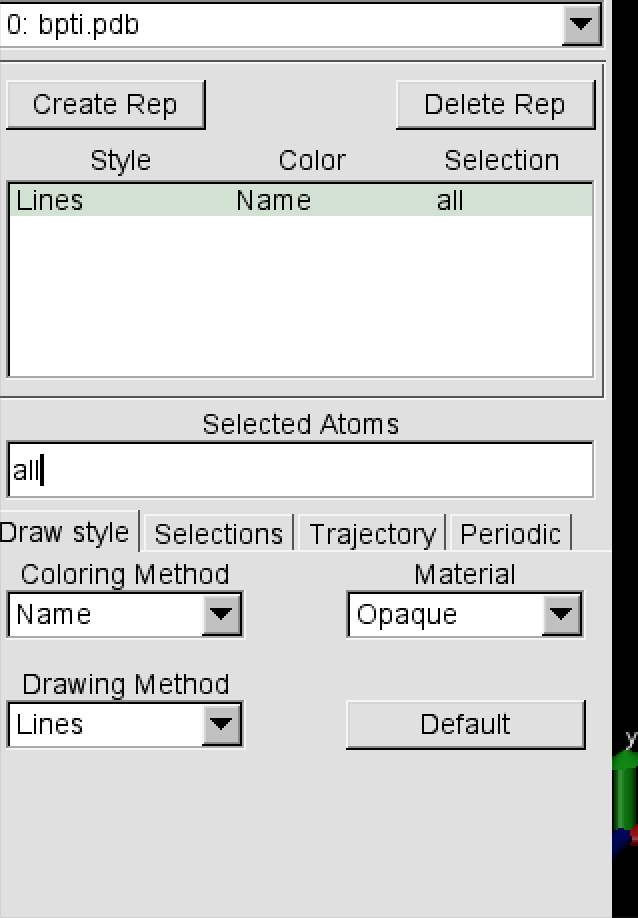
除了默认的显示原则外,我们也可以自定义显示的内容。
在drawing method 中,可以更改为newcartoon,可以非常直观的看到蛋白质的二级结构。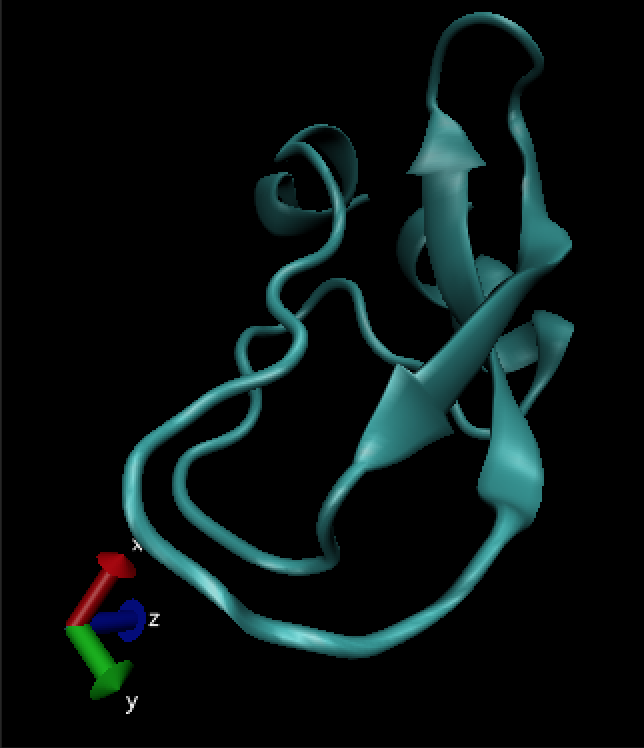
可以为不同的二级结构设立独特的颜色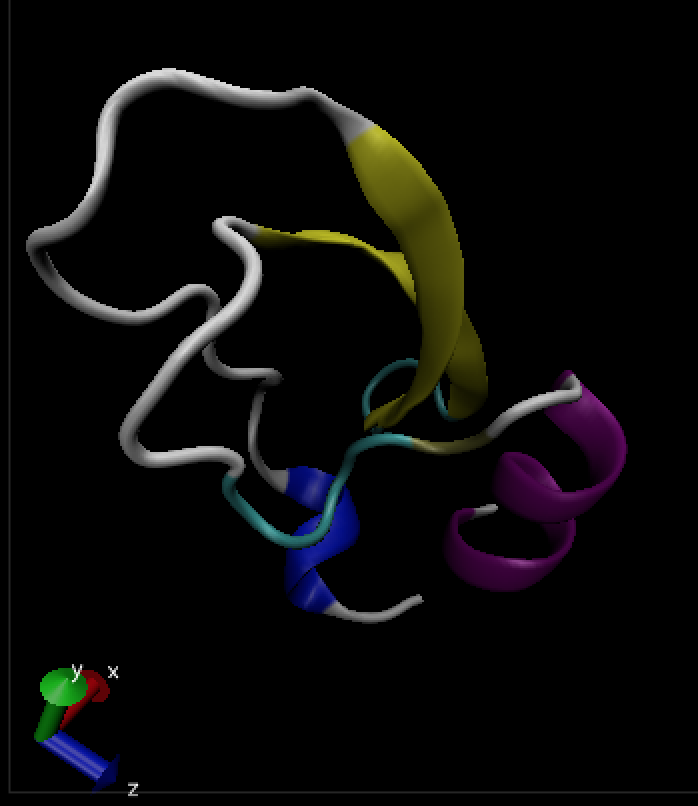
可以根据个人目的选定需要的内容显示(可以借助逻辑词)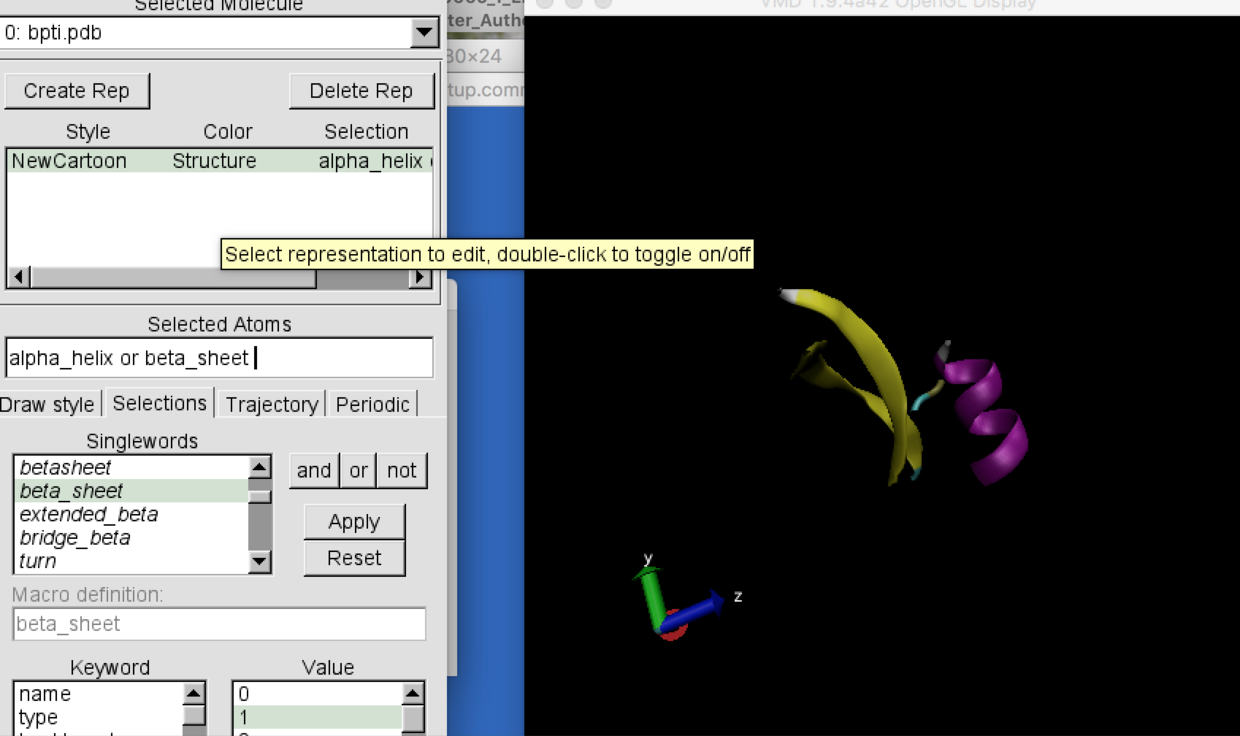
功能非常的多,根据自己的需求可以做各种的调整。(相当不错的一张图了吧~)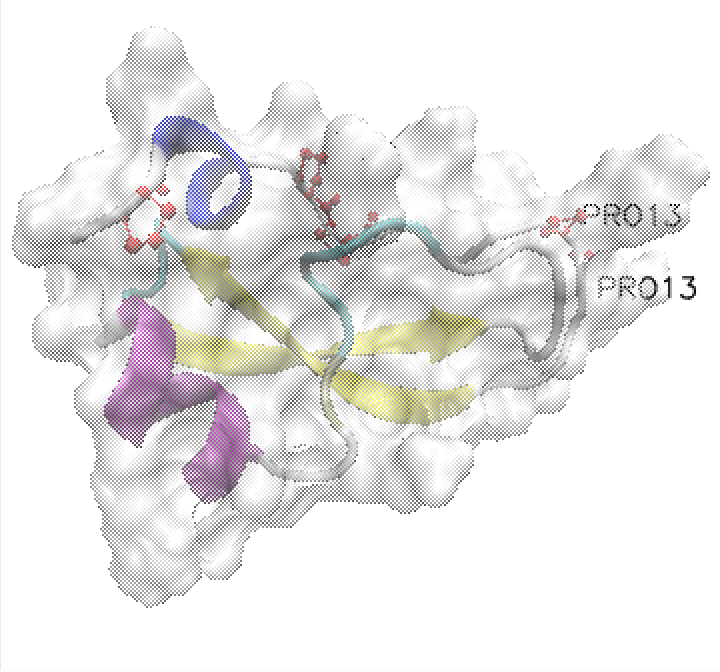
计算方法预测蛋白质的三级结构
同源建模法SWISS-MODEL
原理
相似的氨基酸序列对应着相似的蛋白质结构。
1.首先,找一个与目标序列同源的已知结构作为模版(目标序列与模版序列间的一致度需要大于等于30%)
2.为目标序列与模版序列(可以为多条)创建序列比对。通常比对软件自动创建的序列比对还需要进一步人工校正。
3.根据第二步创建的序列比对,用同源建模软件预测结构模型。
4.评估模型质量,并根据评估结果重复以上过程,直至模型质量合格。
使用swiss model
https://swissmodel.expasy.org/
可以直接使用fasta 格式文件,上传上去后,就可以直接选择build model。或者,也可以自己查看模版,自定义比对中选择的模版链。
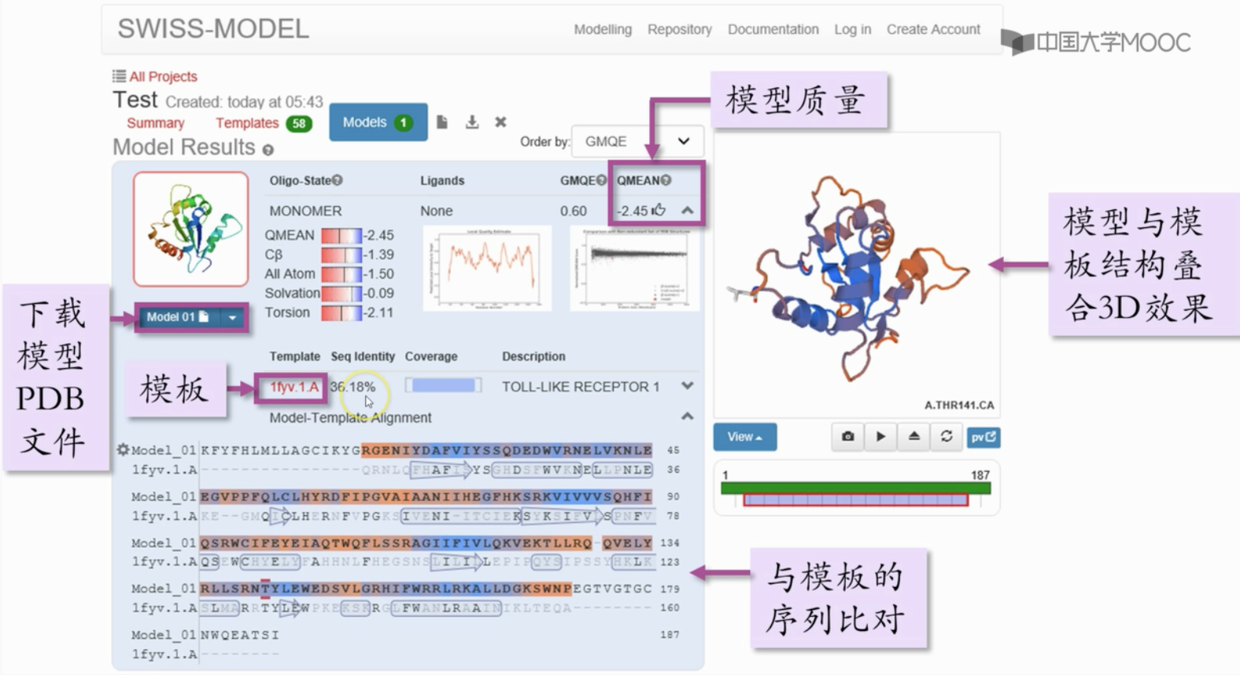
预测结果
如果目标序列与模版序列一致度极高,那么同源建模法是最准确的方法。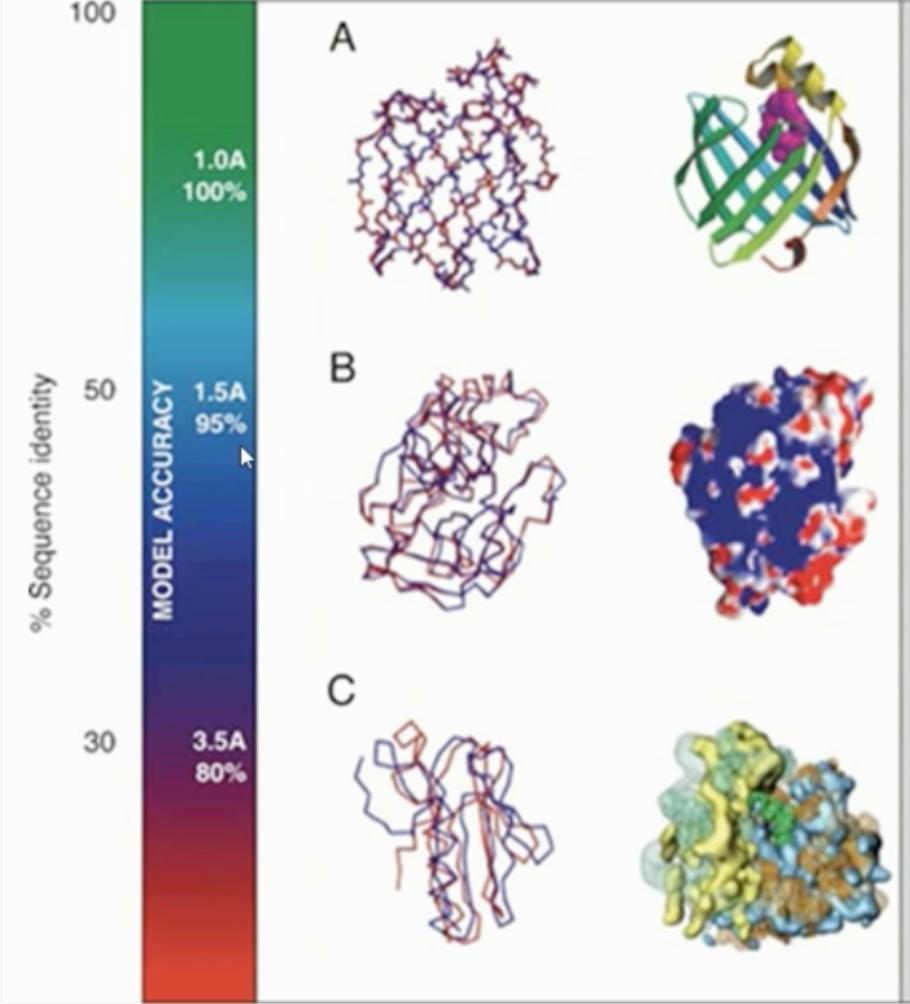
- 当然也存在特殊的情况,比如序列的一致度虽然很高,但是结构却并不相同。
- 另外,如果目标序列与模版序列一致度
小于30%,则这个方法不适用。
穿线法 I-TASSER
原理:不相似的氨基酸序列,也可以对应着相似的蛋白质序列。
(这点是不是和同源建模很不一样?)其实也就是互为补充啦。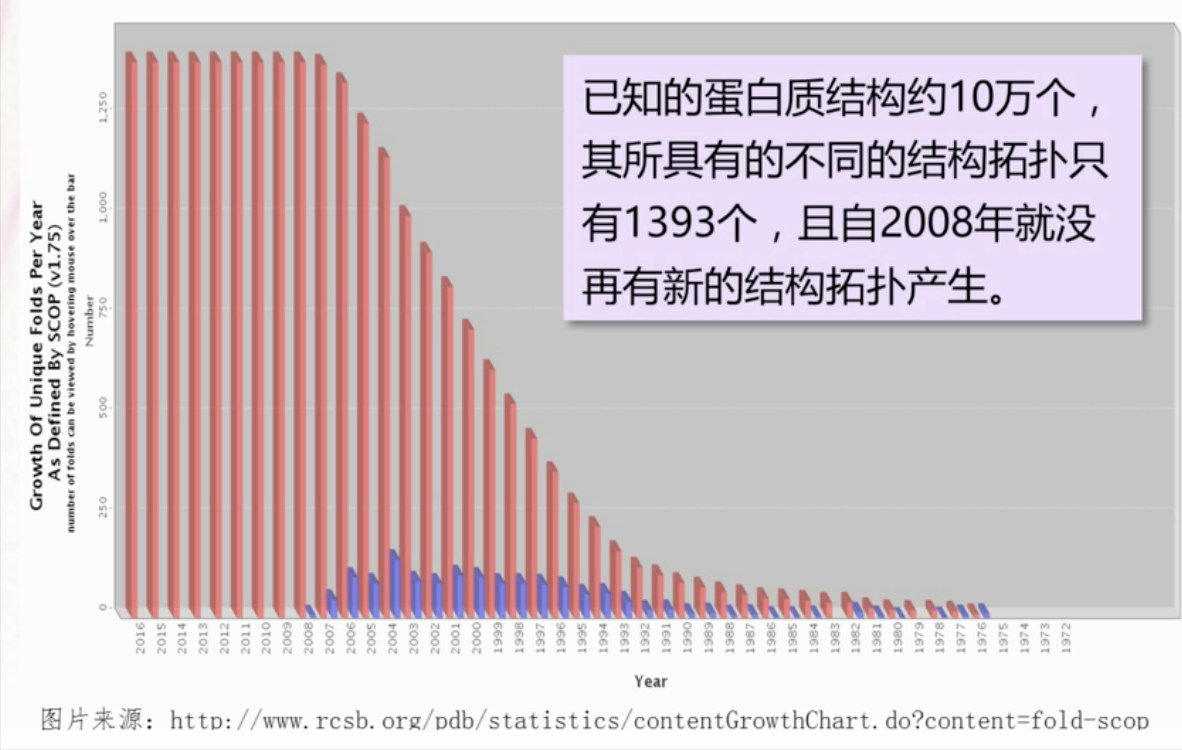
我们可以将目标序列,像“线”一样,穿到现有的结构里,看哪个结构最终结果最合适,哪个结构就作为模版。这个结构是否合适,用能量方程来评定,能量方程越低,就越合适。
使用I-TASSER
https://zhanglab.ccmb.med.umich.edu/I-TASSER/
该网站使用学术或办公邮箱注册就可以使用了。
一个IP 一次只能提交一个任务,而穿线法需要的时间比同源建模法也要长许多,一般都需要十几个小时!
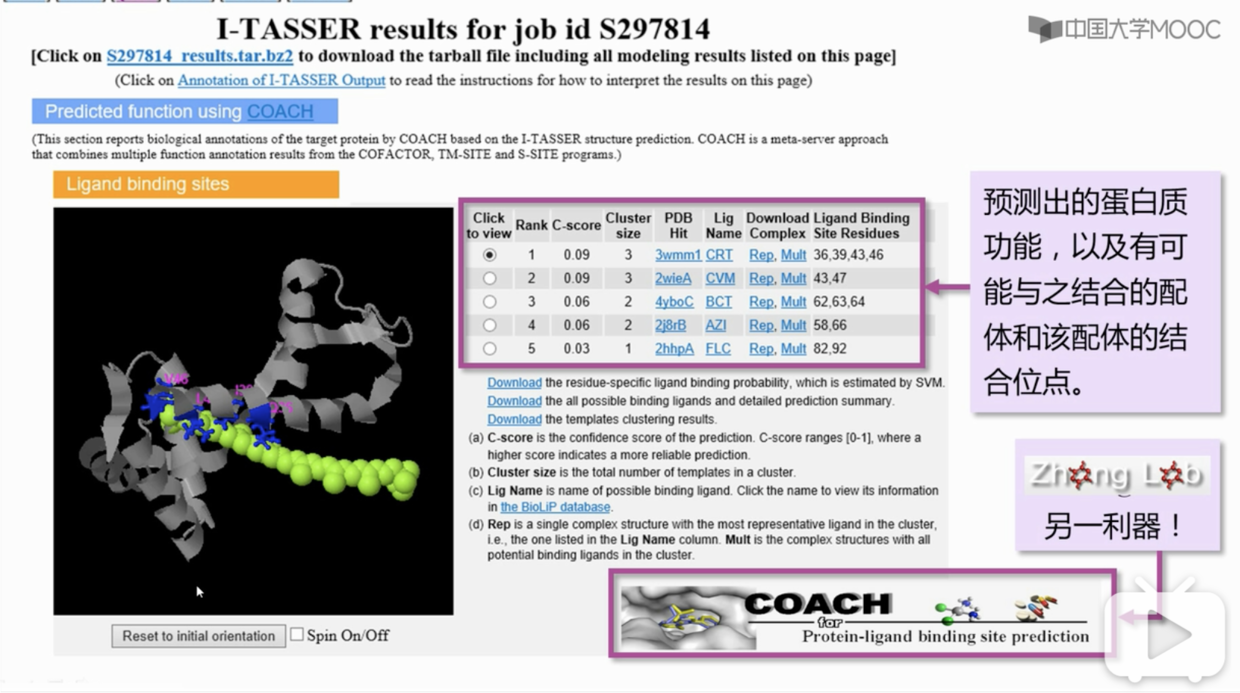
- 结果
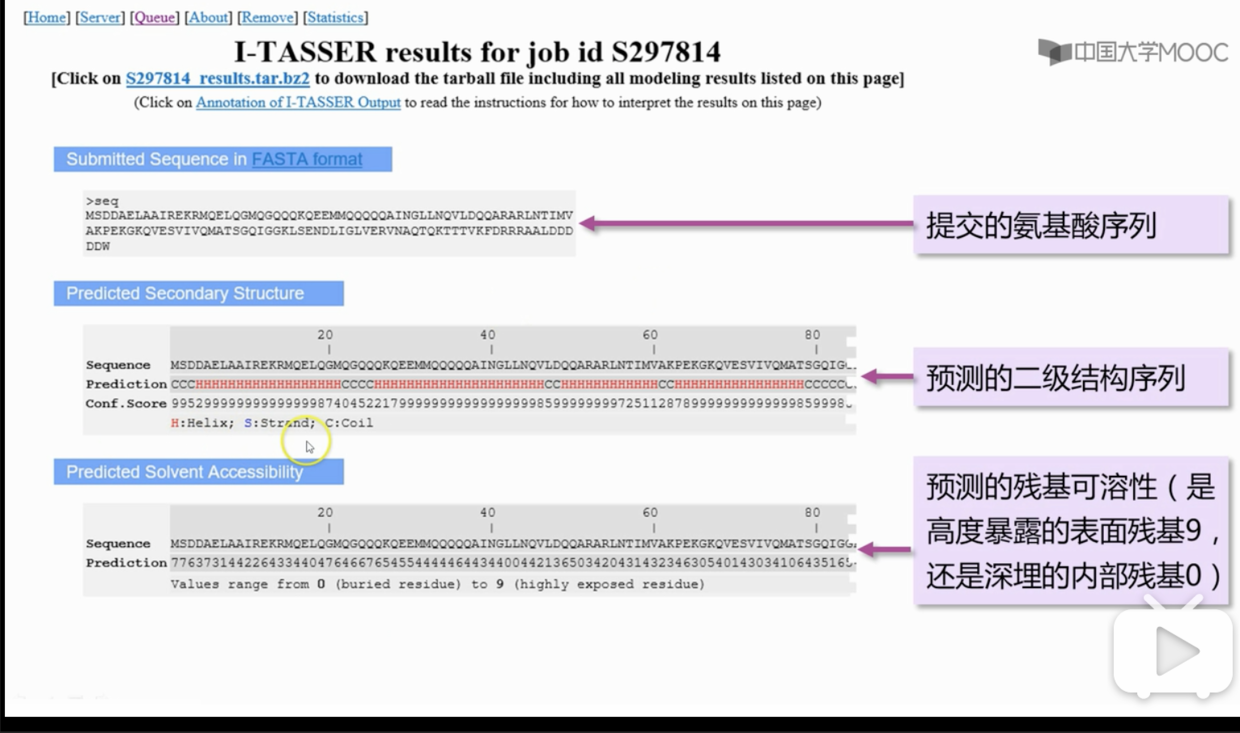

I-TASSER 给了多个预测模型,主要看TM-scores,要求在0.5 以上最为可信。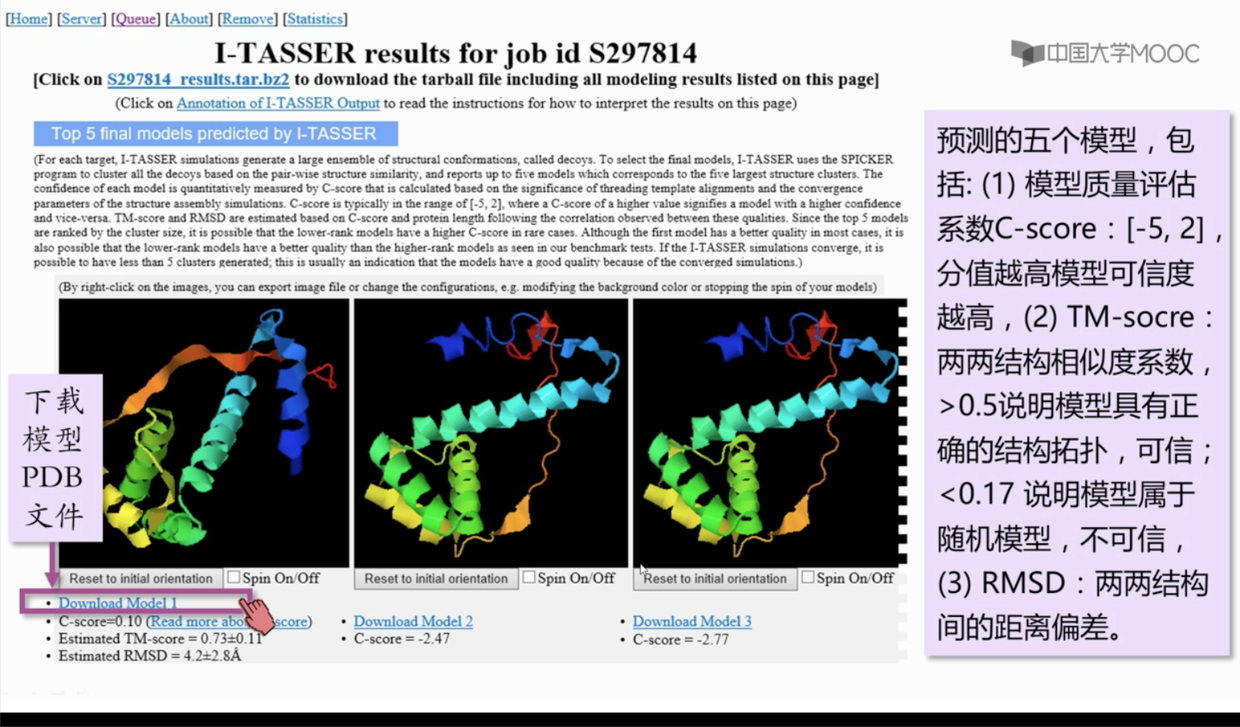
- I-TASSER 还能找出与得分最高的结构最相似的十个蛋白结构,用以对其
功能进行推测。
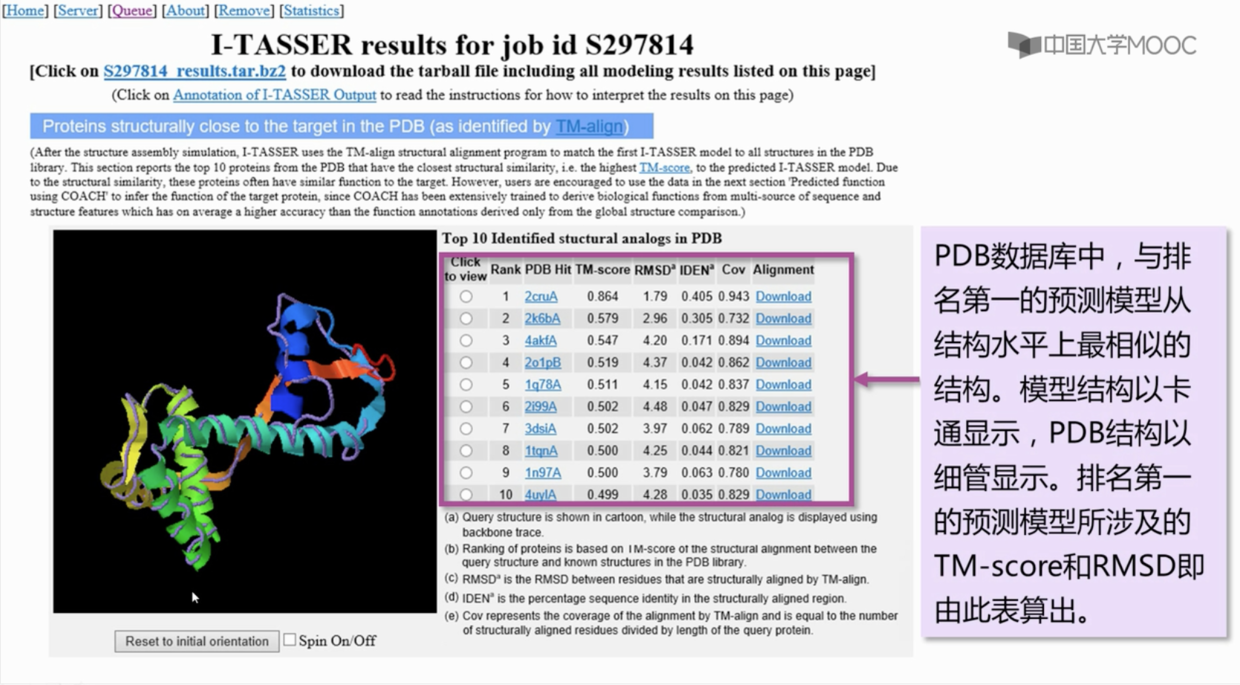
- 甚至还给出了可能的配体以及配体的结合位点(使用的是另一个软件的技术
COACH)
从头计算法 QUARK
有的时候,可能穿线法也无法解决三级结构的预测问题。比如预测出得模型评估系数都不合格。
“蛋白质的三维结构决定于自身的氨基酸序列,并且处于最低自由能状态。”(长成自由能最低的情况,最稳定)
从头计算法就是预测氨基酸所有的可能的三级结构,并计算它们的自由能,选择自由能最低的那个结果。(这工作量~)
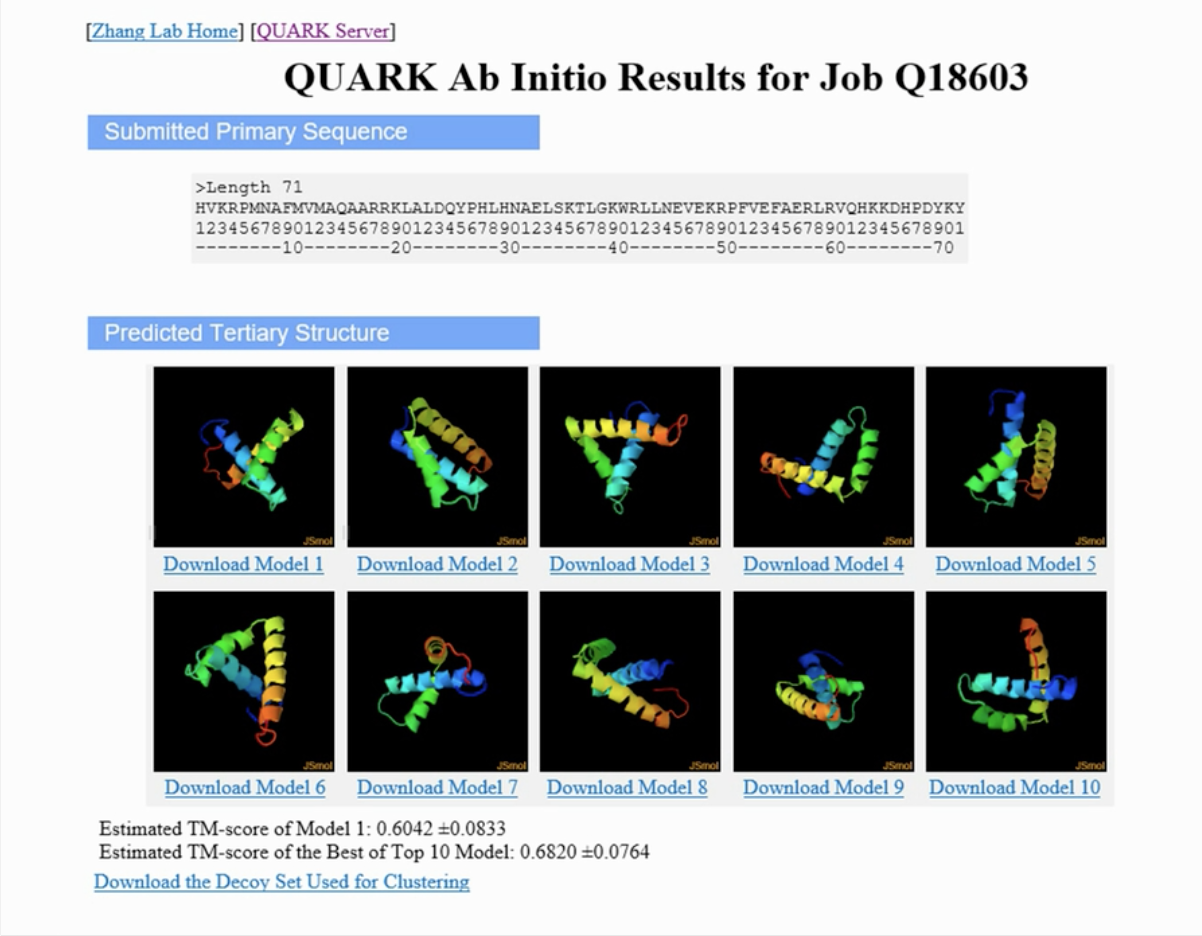
- 依旧是Zhang lab 的软件,quark(只能预测氨基酸序列在200以内的)
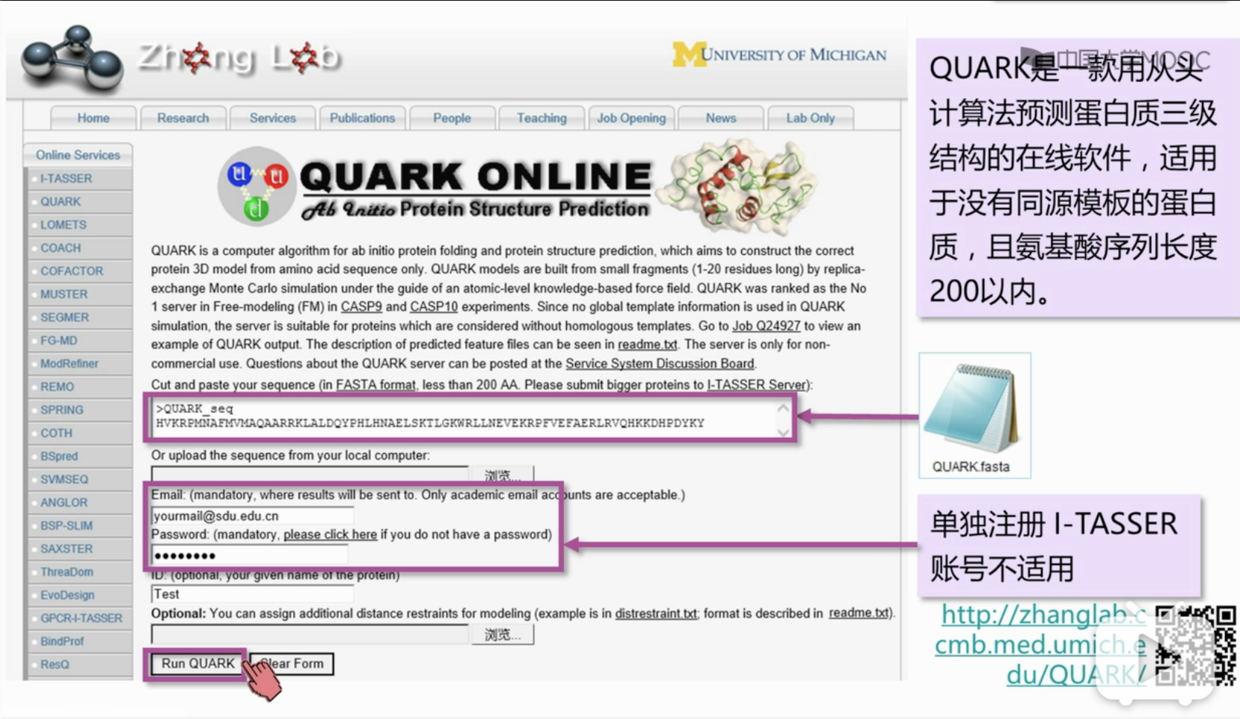
- QUARK 的计算量也决定了它的时间的消耗,一般需要2d 以上(等吧~)
还是主要以TM-score对模型进行选择
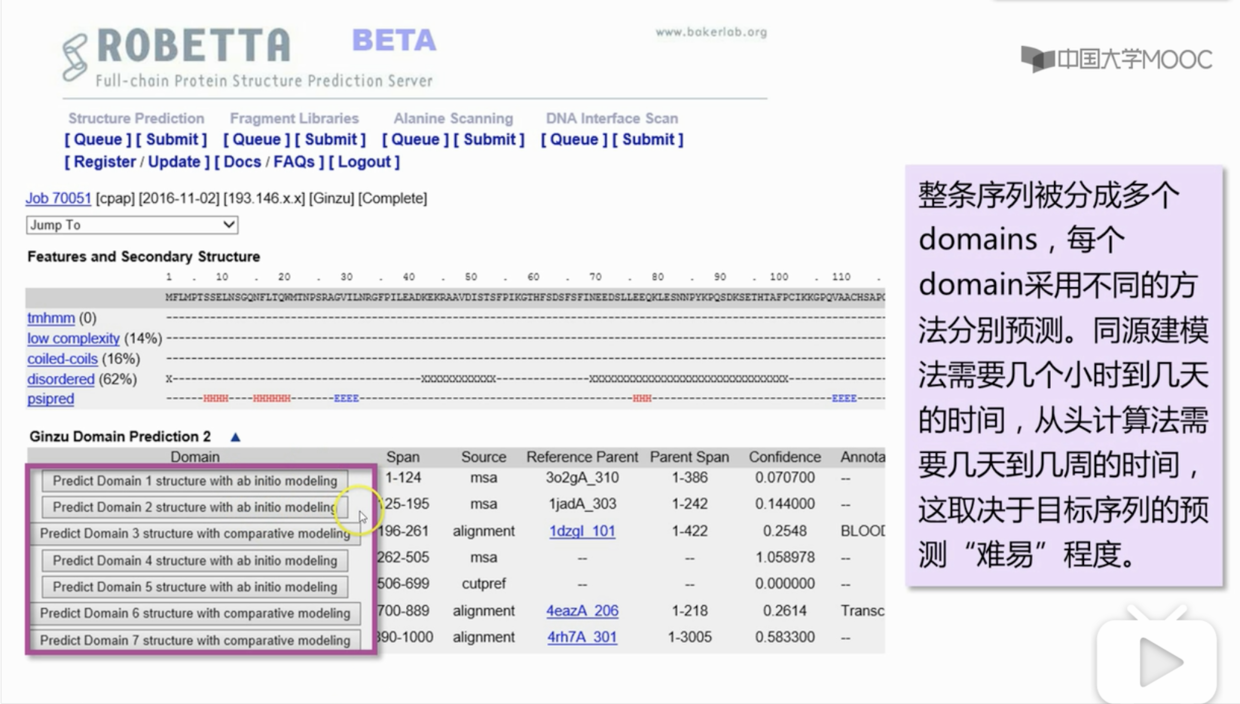
综合法 ROBETTA
大合一大法好~
原理:综合法直接综合了同源建模法、穿线法与从头计算法等的两种或多种,将氨基酸序列进行分段,情况不同的片段采用不同的方法。
ROBETTA
ROBETTA 结合了同源建模法与从头计算法这两种方法,能找到模版的区域会使用同源建模法,而找不到模版的区域,则使用从头计算法。
http://robetta.bakerlab.org/
- 不仅比对预测需要等,这个排队竟然还要等!

ps:确实是了不起的项目
https://boinc.bakerlab.org/rosetta/
- 使用robetta 预测更加需要耐心
总结
- 到底该用谁呢?
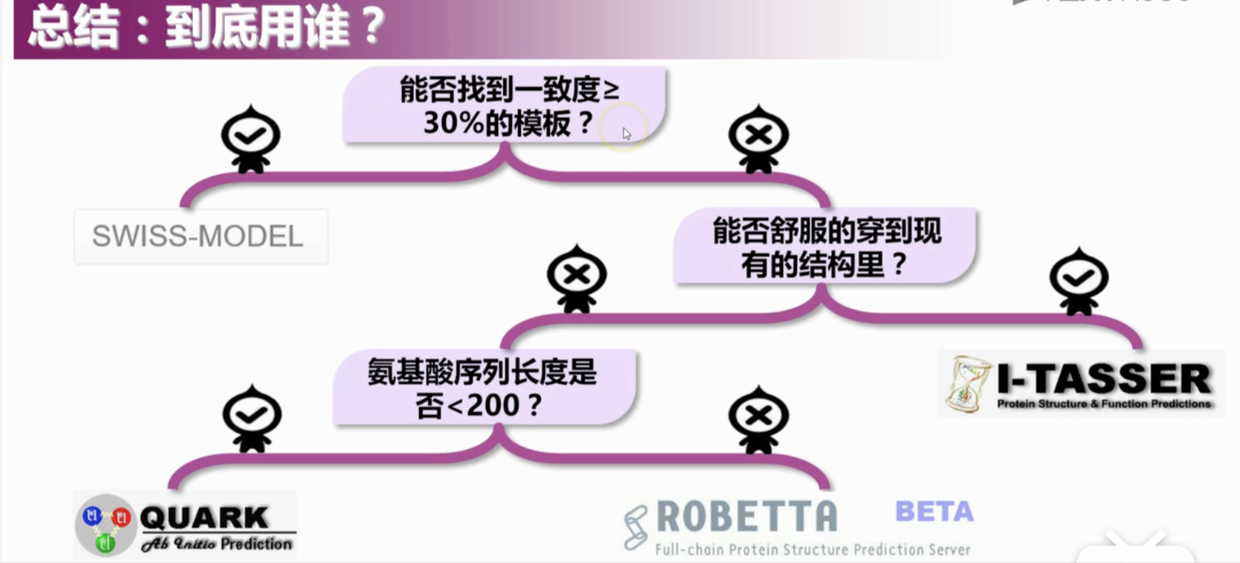
模型质量评估

- 大部分的结构预测软件自身就带有模型评估的功能
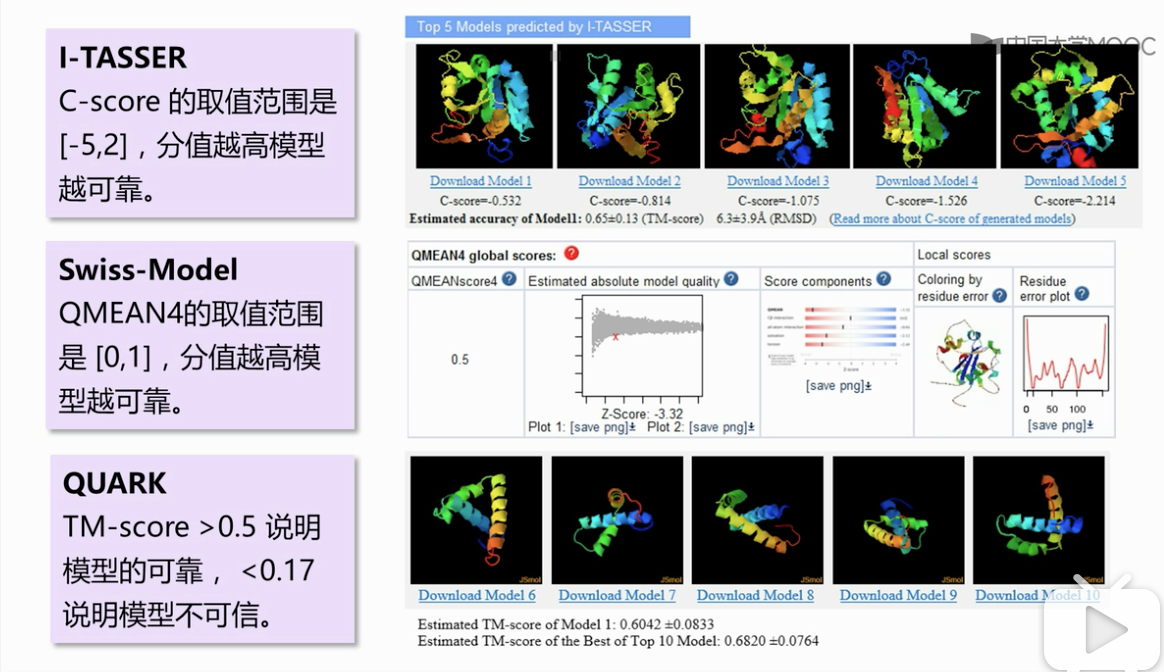
- 但一般来说,模型必须得至少从
三个角度进行验证,起码三个合格,才能评估为可靠的模型。
SAVES v5.0 评估平台
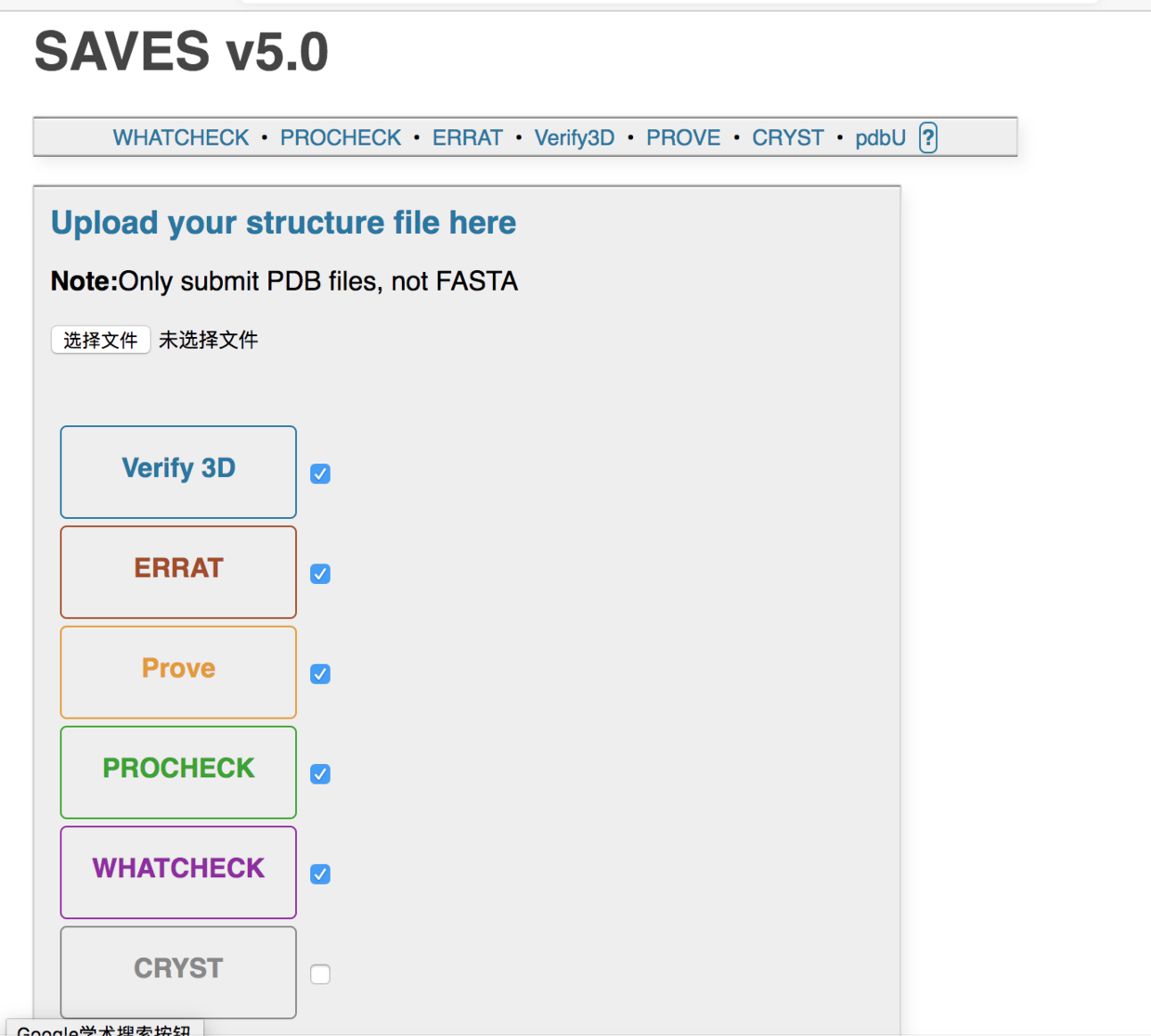
SAVES 平台提供了六个软件。
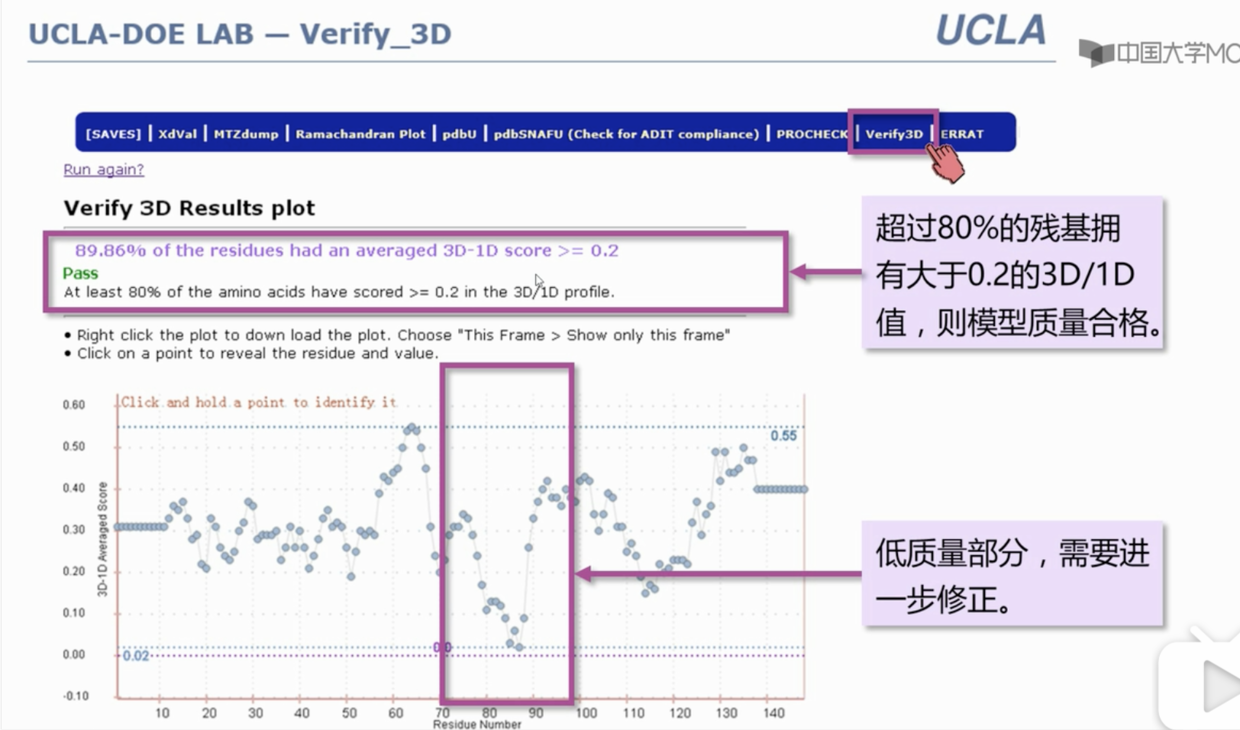
verify_3D
PROCHECK
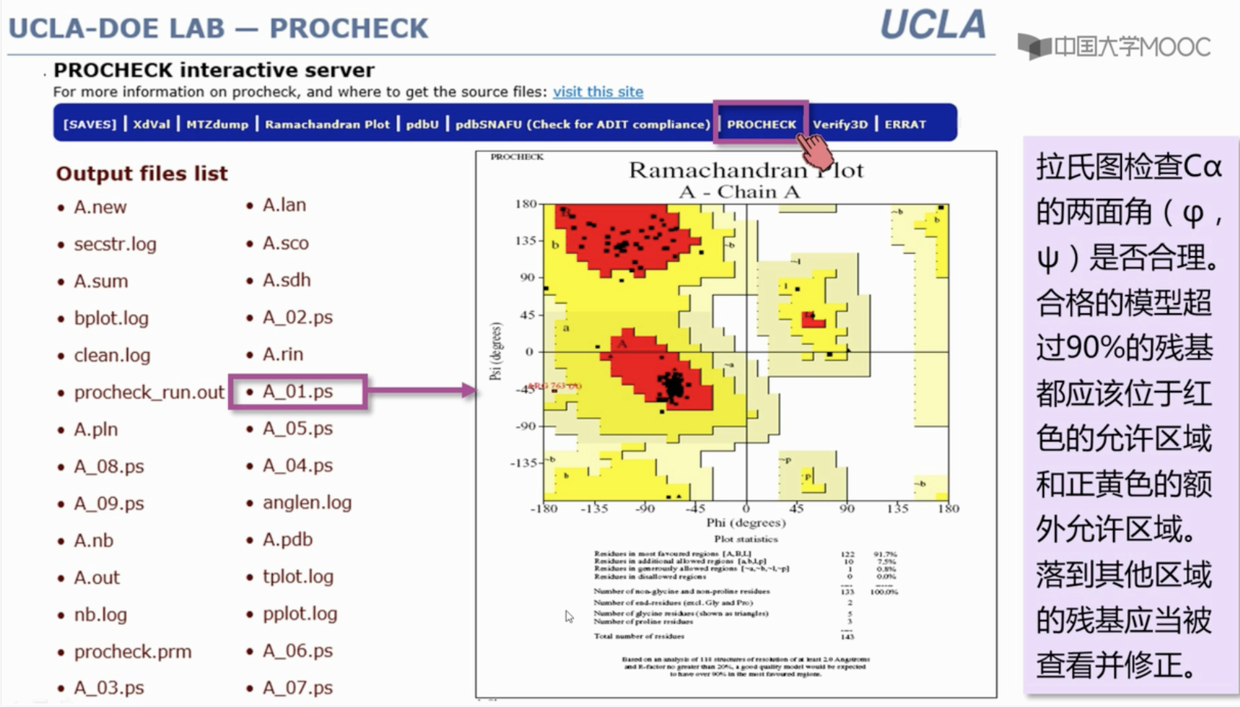
ProQ
proq 也是一个不错的评估平台。
http://www.sbc.su.se/~bjornw/ProQ/
ps:目前访问崩了~
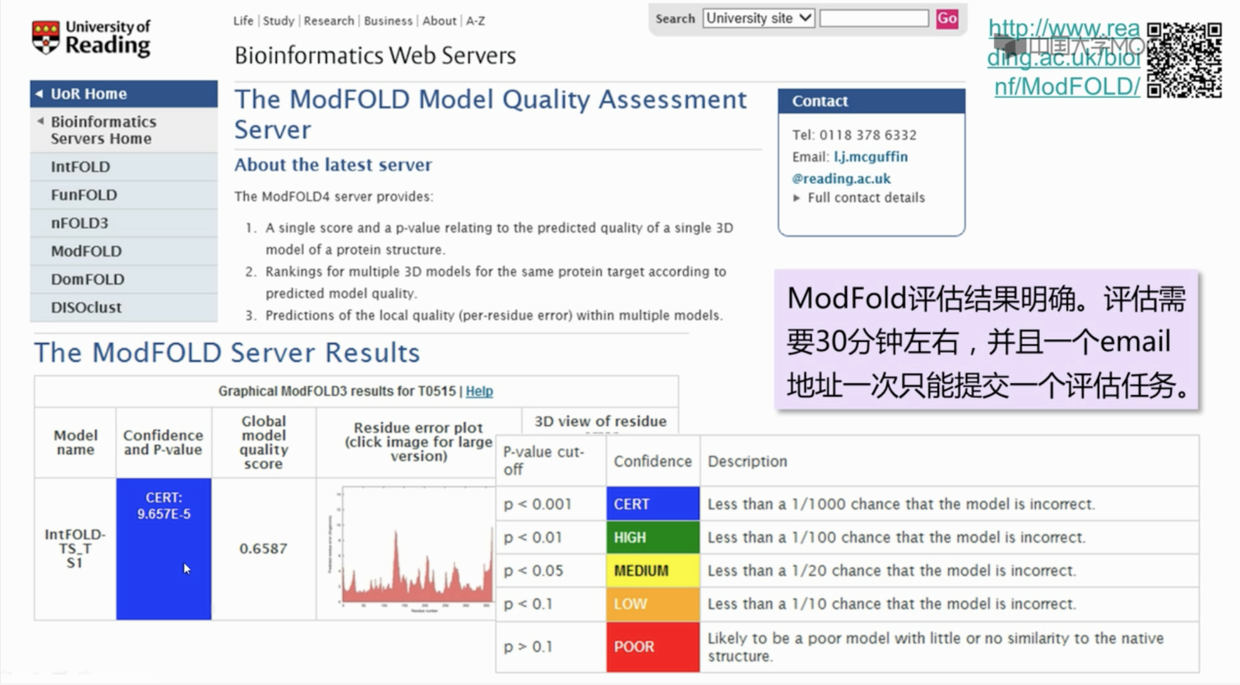
modfold
三级结构的比对
结构比对,就是对蛋白质的三维空间结构的相似性进行比较,是蛋白质结构分析的重要手段之一。
1)可用于探索蛋白质进化及同源关系;
2)改进序列比对的精度;
3)改进蛋白质结构预测工具;
4)为蛋白质结构分类提供依据;
5)帮助了解蛋白质功能。
superpose 平台
http://superpose.wishartlab.com/
- 直接将需要进行比对的pdb 文件添加即可。

SPDBV 蛋白质结构分析平台
SPDBV 是一款用于蛋白质同源建模的软件。可进行整体智能叠合,或者选择性叠合。
对于mac,10.11 以后的版本,需要通过ftp 下载。(我是直接使用wget)
- 界面如下,直接两次打开两个需要进行比对的pdb文件即可。
- (先对color 设置layer,区分二者颜色)在fit 选项中使用magic fit,选择all atoms。
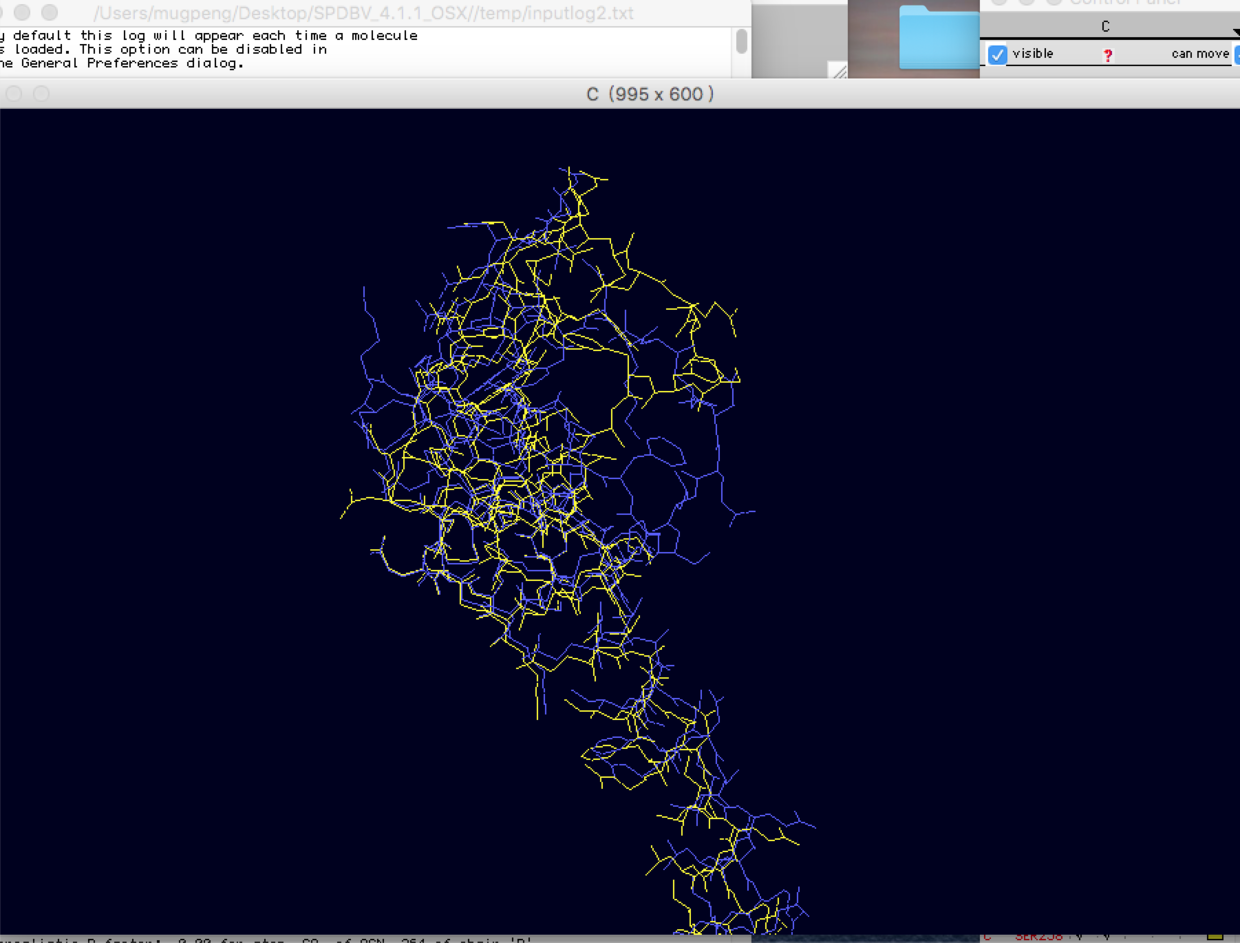
- 在control panel 中,分别对C、D(两个比较的蛋白)进行操作:选中所有的氨基酸,再取消选中258-281 号氨基酸。把结构不同的区域排除在外。(两个选中的蛋白的氨基酸序列的个数必须一样多)
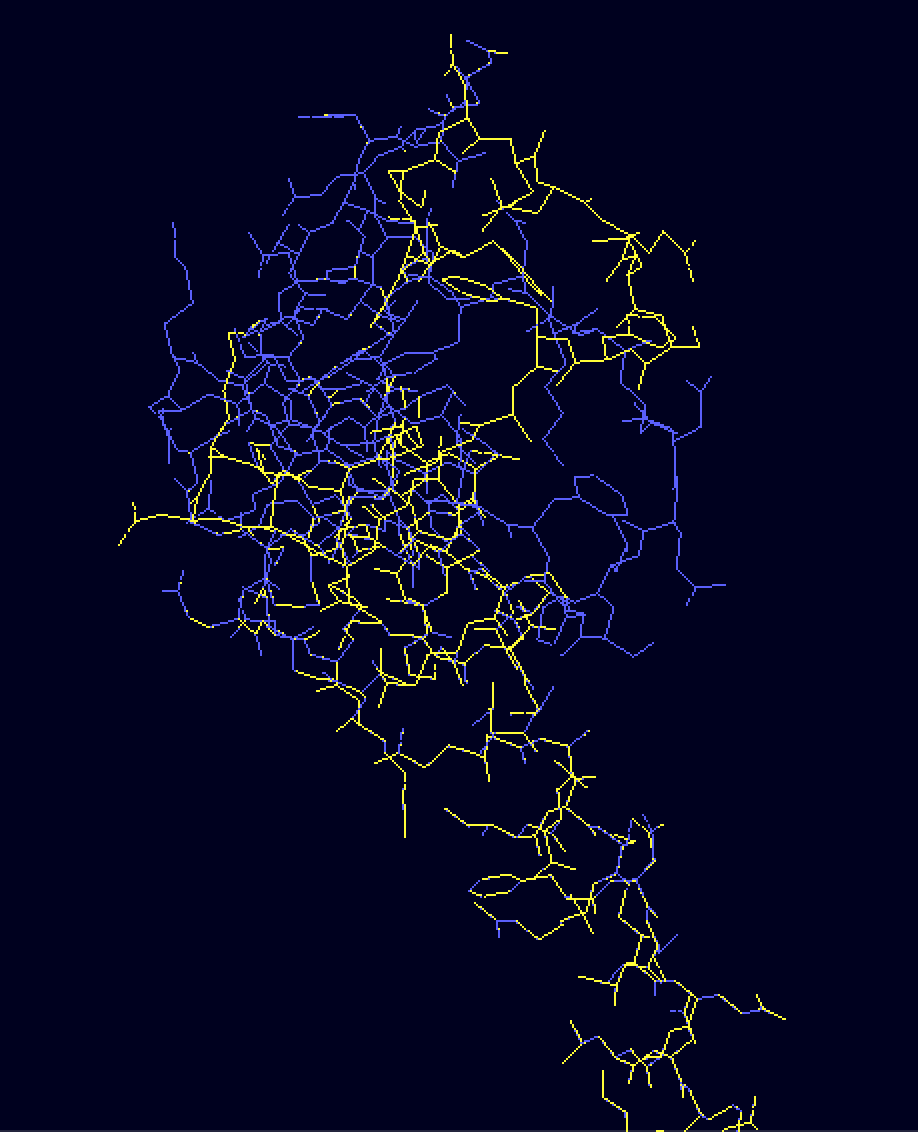
蛋白质分子表面性质
1)表面形状;(vmd,surf representation)
2)表面电荷分布;
3)表面残基可溶性;
表面电荷分布
我们可以通过VMD 软件的插件,APBS,制作表面电荷分布。
- 可以在APBS 官网下载文件,并将该文件拖拽到VMD 软件的目录下。
https://sourceforge.net/projects/apbs/ - 导入pdb 文件,并将文件内容转换为psf 文件。(APBS识别)
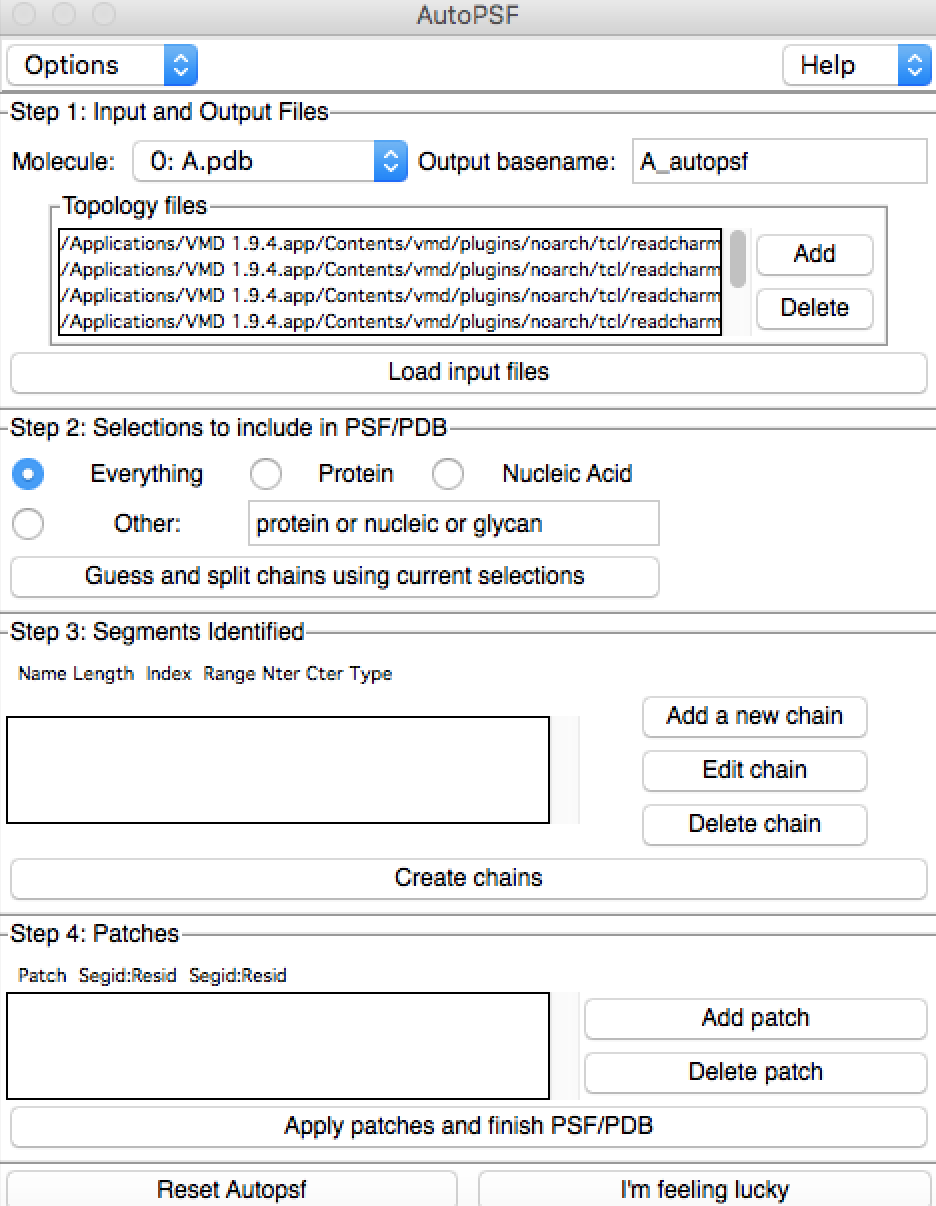
- 后面需要load 文件,接着guess 选项,再最后选择
l'm feeling lucky。 - 完成之后,会在电脑工作目录下(也可以自己设定为指定目录)创建psf 文件。
- 重新打开vmd。先载入新生成的pdb 文件,再载入psf文件(将psf 加载至pdb 文件之上。)
- 在extension 中选择APBS tools。将APBS 文件指定到输出位置。
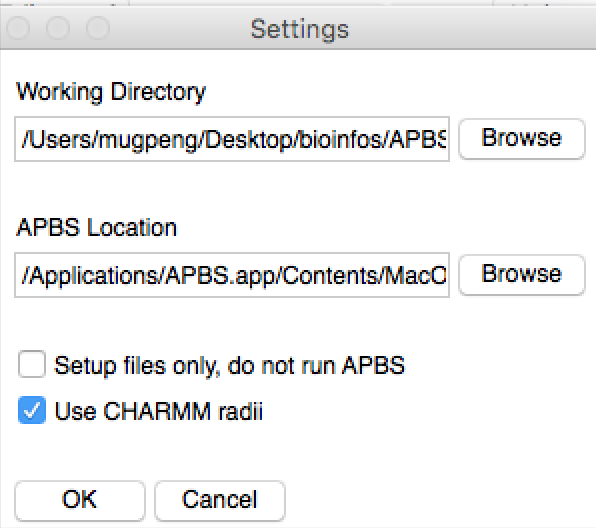
- 开始run 就完事儿啦
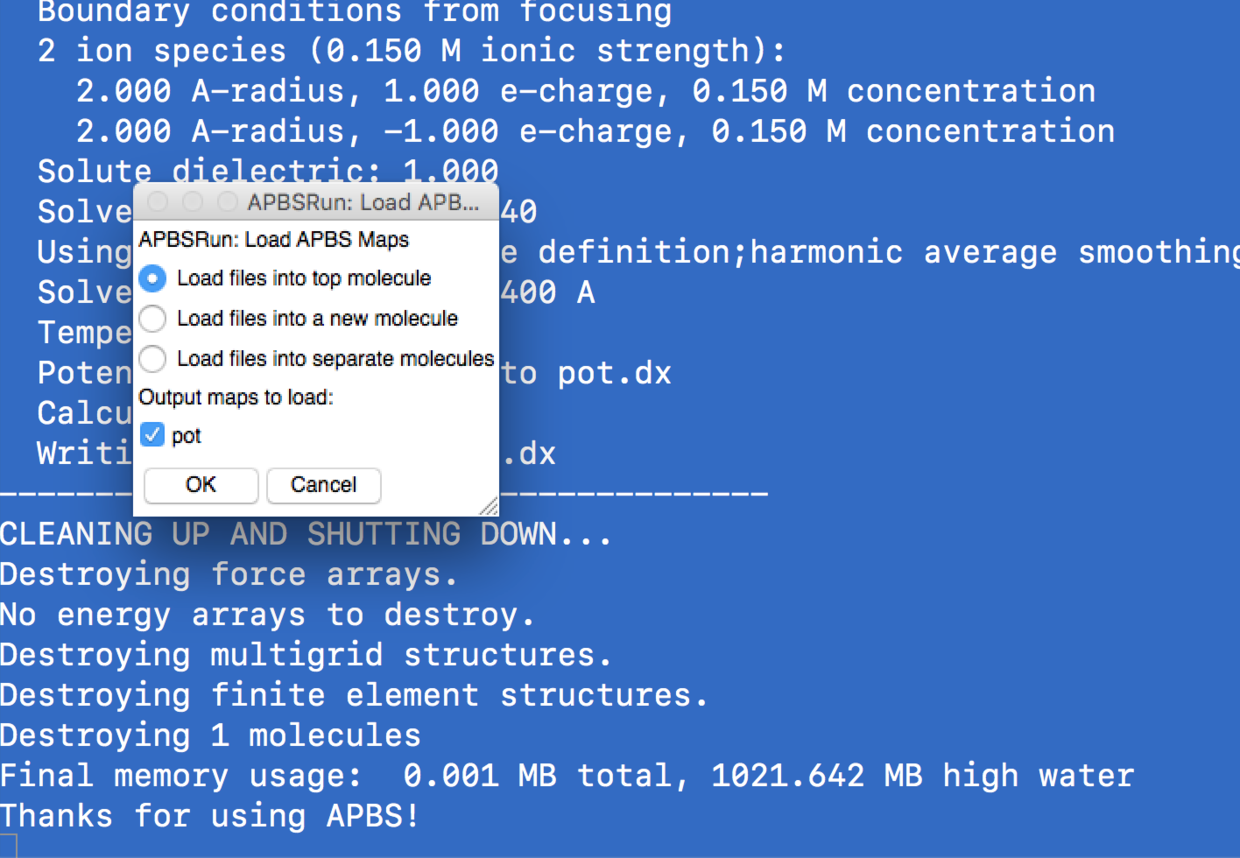
- 重新打开vmd。分别载入
pdb, (之后文件都是载入pdb 文件之上)psf,pqr,dx。 - 调整color method 为volume,drawing method 为surf,选择trajectory,调整为-10-10。就可以看到电荷分布了。
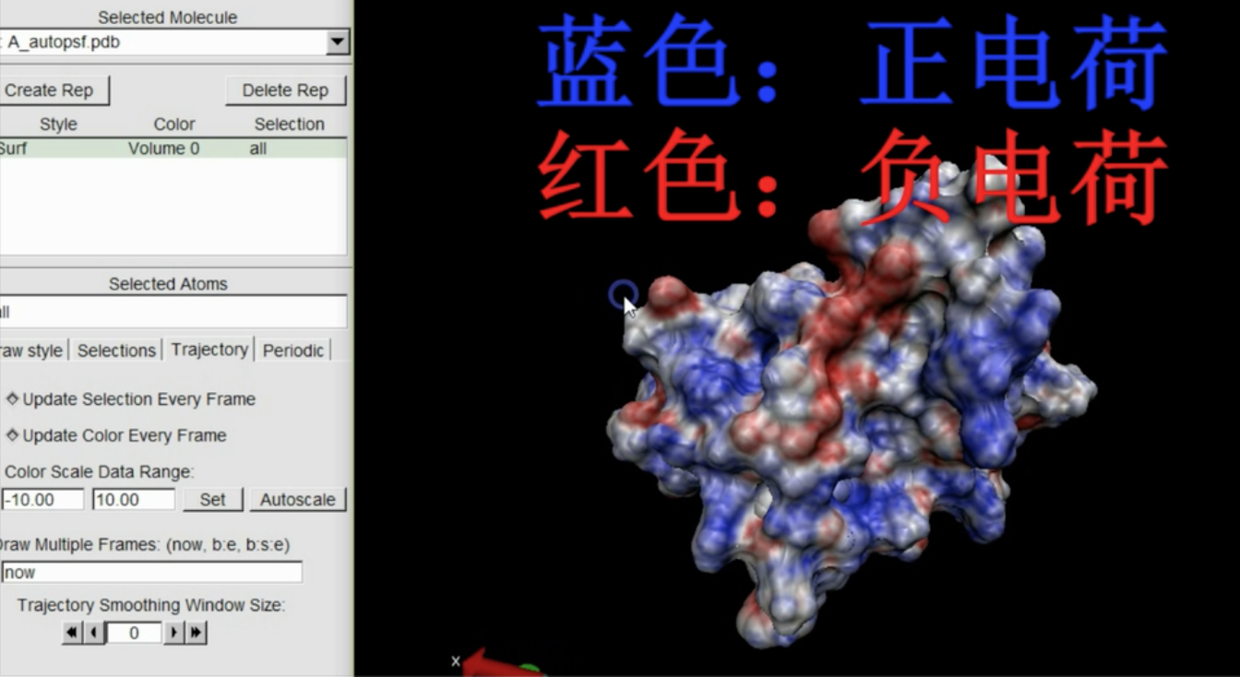
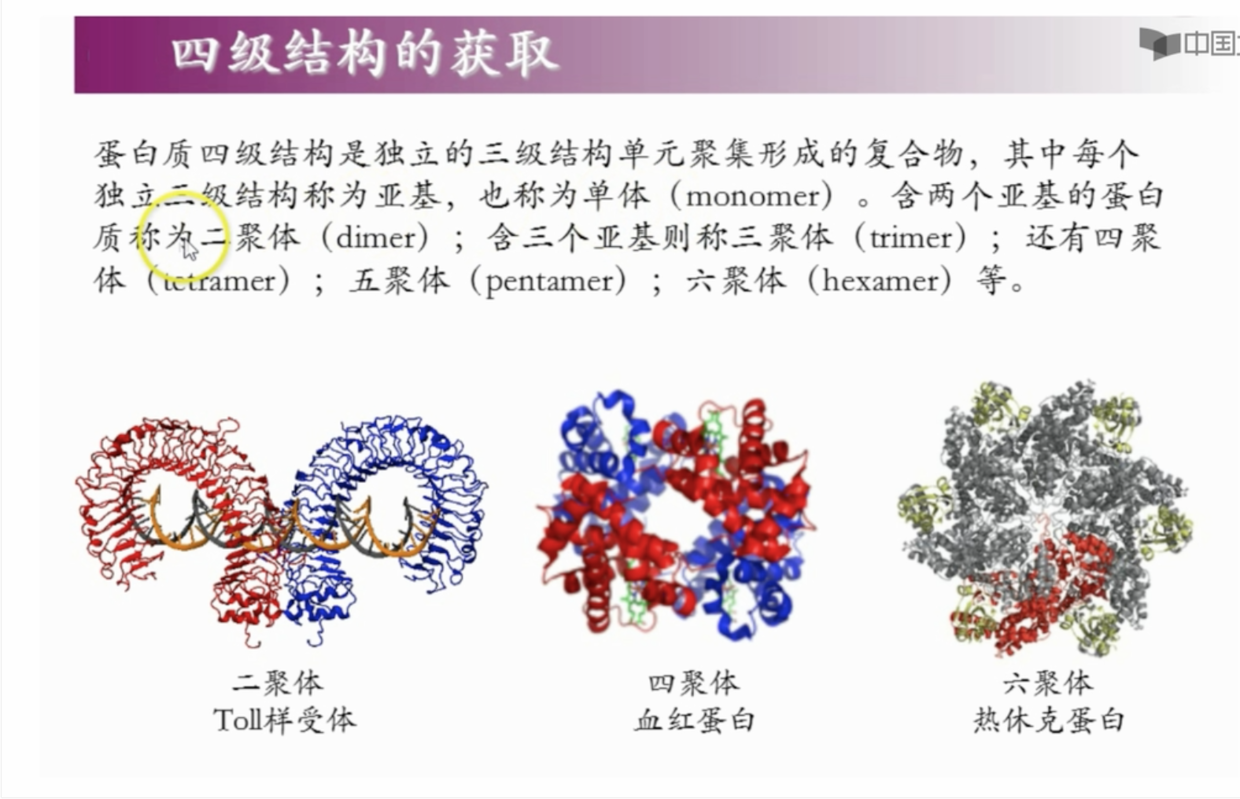
蛋白质四级结构
- X 射线衍射法
- 冷冻电子显微技术


若有收获,就点个赞吧
0 人点赞
