分子对接
分子对接计算
配体分子放在受体活性位点的位置,然后按照几何互补,化学环境互补与能量互补的原则评价配体和受体相互作用的好坏,并找到分子之间最佳的结合模式。
从大量的化合物数据库中的化合物逐一与靶标分子进行对接(docking),通过不断的优化小分子化合物的位置(取向),以及分子内部柔性键的二面角(构象),寻找小分子化合物与靶标大分子的最佳结合构象,并计算二者的相互作用和结合能。(筛选排名靠前的化合物)
分子对接的结合强度取决于结合的自由能的变化。
我们可以设定相关的阈值,比如rmsd,作为筛选的标准,从而减小化合物库中化合物的数量。
打分函数
- 基于经验的回归参数;
- 基于分子力场的方法;
- 基于知识的打分函数。
discovery studio-PLP1;
sybyl -chemscore;
gold-asp。
数据库
pdb 蛋白结构数据库,可以参考:https://www.yuque.com/mugpeng/bioinfo/nrldxt#e6ecb442
蛋白质-蛋白质 分子对接
我们可以通过分子对接,docking,技术对蛋白质四级结构进行预测。
分子对接会尝试所有可能的结合形式,并根据打分函数给每种形式打分排名。(能量越低越稳定)
对接过程会考虑如下因素:
- 形状互补
- 亲疏水性
- 表面电荷分布
一共有两种蛋白质-蛋白质分子对接方式:
rigid docking 刚性对接:目前可用的大多数软件为刚性对接。
flexible docking 柔性对接(蛋白质真实状态):计算量大,可用软件少,且多为收费。
使用zock 进行分子对接
- 需要学校邮箱(但好像有的学校不在名单中无法使用)

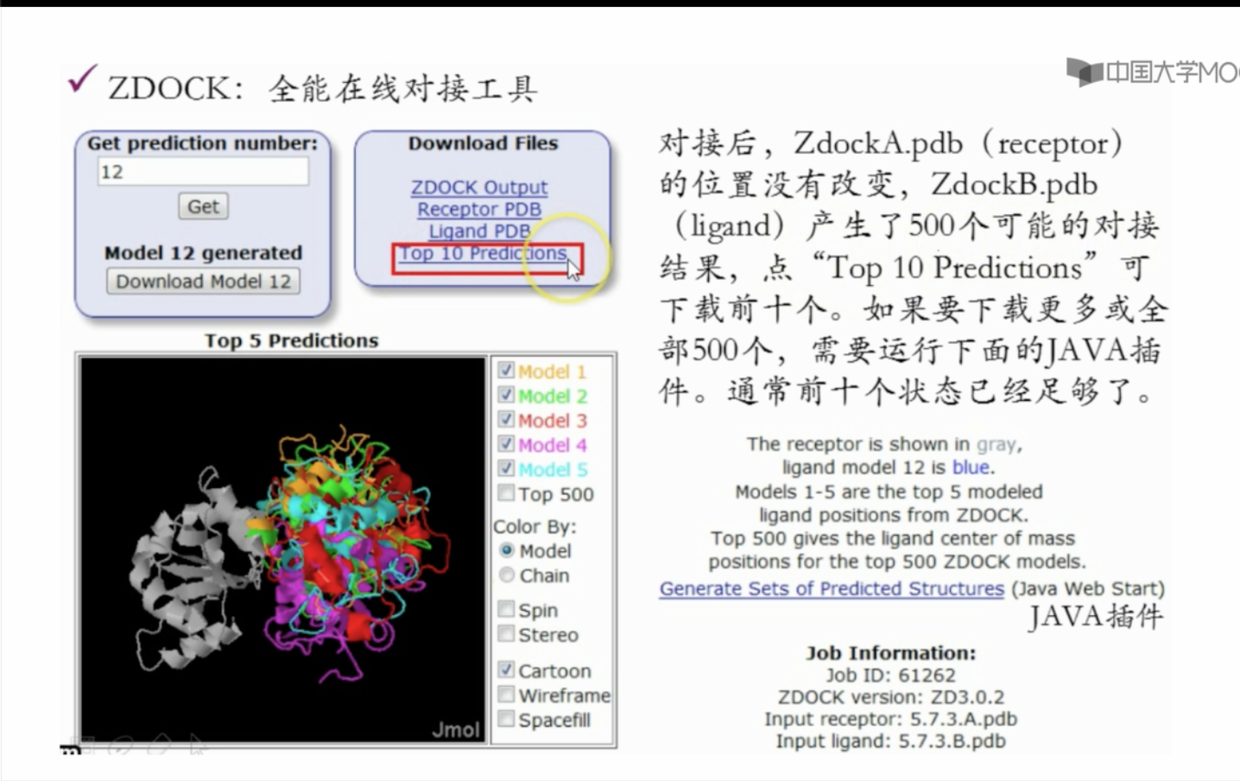
http://zdock.umassmed.edu/ - 可以进一步选择
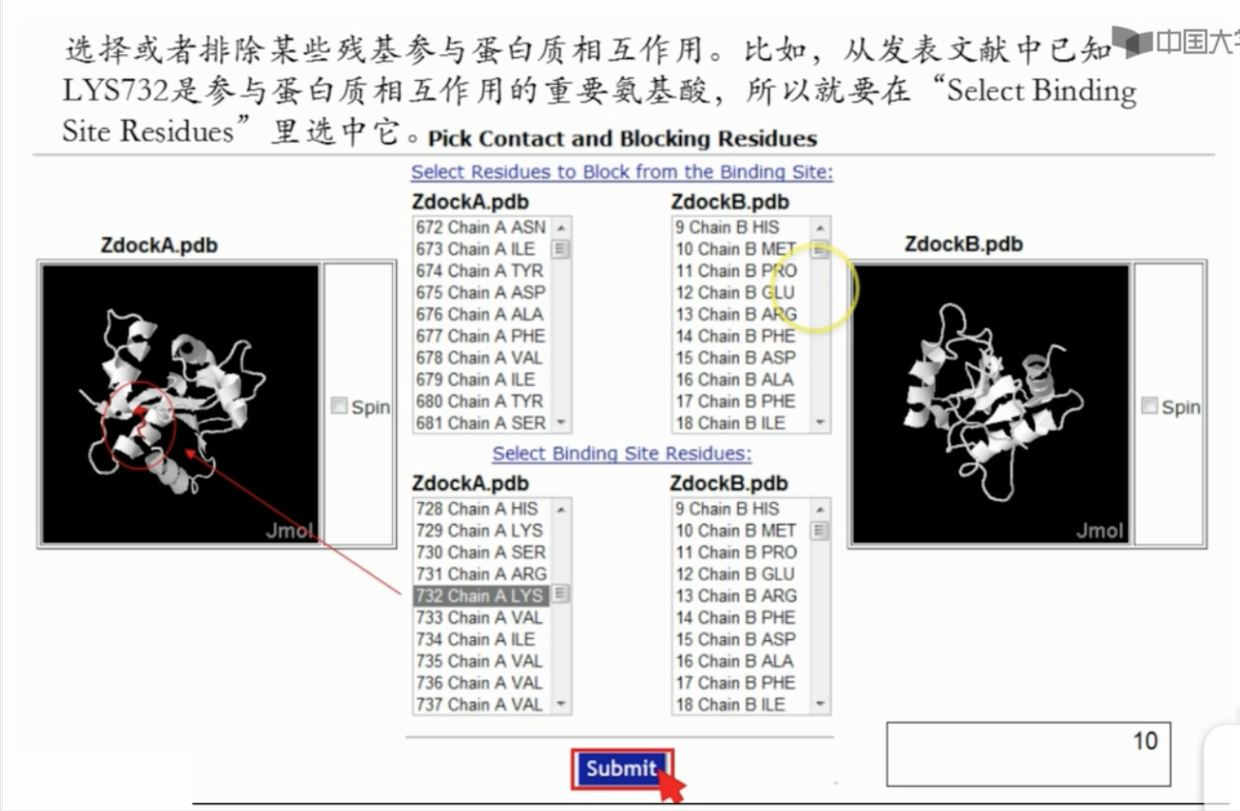
- 大约30min
PDBePISA 进行蛋白对接分析
可以对分子对接比较后的结果进行分析。
- 可以在interface 界面下查看相关的信息
自动能结果小于零才有意义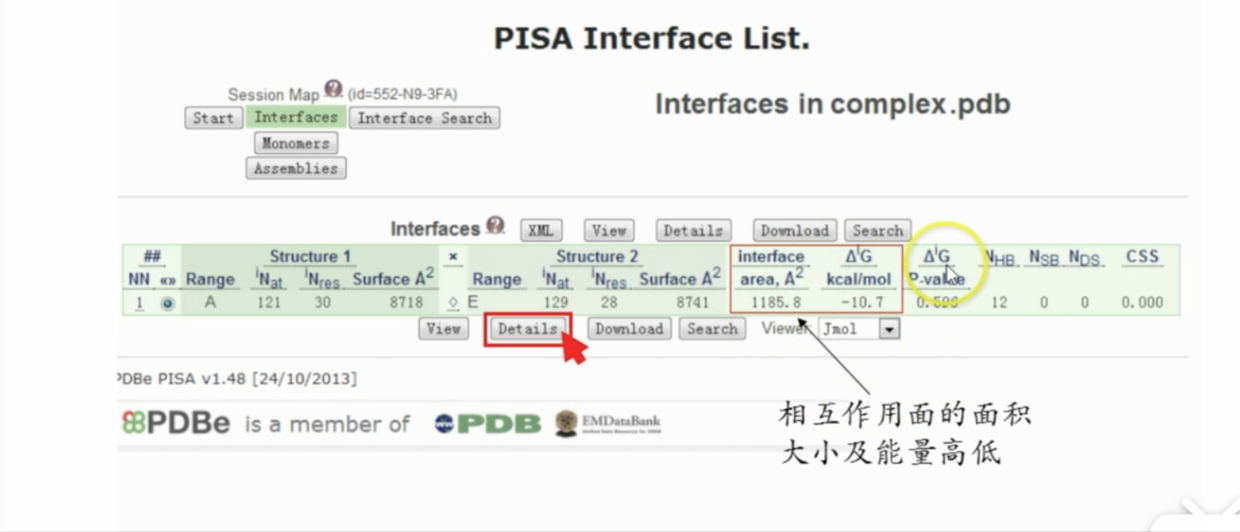
- interfacing residues 界面记录了所有参与相互作用的残基
蛋白质小分子对接
- 刚性对接:靶标和配体始终保持刚性,也就是说研究体系的构象不发生变化;
- 半柔性对接:研究体系中的靶标和配体允许在一定范围内变化;
- 柔性对接:小分子和靶标在对接过程构象可以自由变化。
使用autodock和MGLtools进行小分子对接
http://autodock.scripps.edu/
autodock 并没有设计图形化界面(貌似mac 是的),因此需要我们下载相关的软件,手动移动到工作目录后用命令行打开。(还需要python2 的开发环境)
相关安装可以参见
http://autodock.scripps.edu/downloads/autodock-registration/autodock-4-2-download-page
接着安装mgltools
http://mgltools.scripps.edu/downloads
记得选择匹配自己cpu型号的安装包(intel)
mac 需要配备xcode 11才可以打开!
- 准备好ligand 与protein 文件
- 打开autodock 后,首先载入ligand 文件。
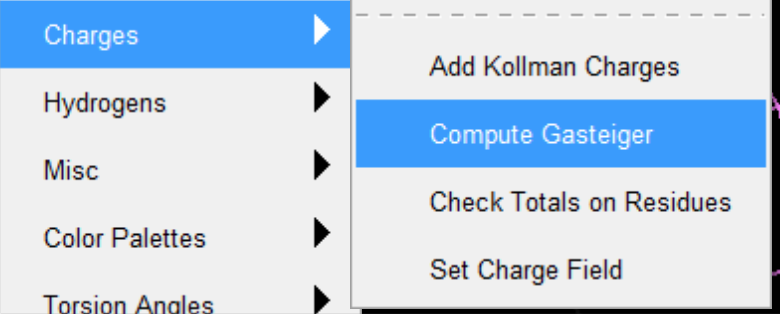
- 为ligand 添加氢键和电荷。
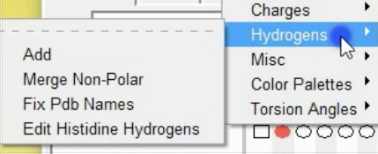
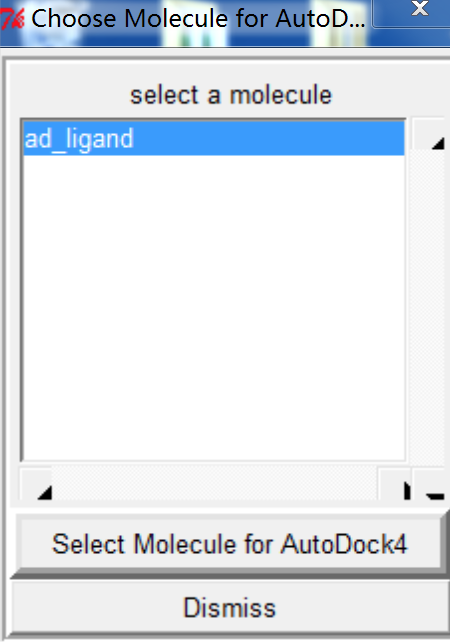
- 选中对接的配体
- 找出配体中心
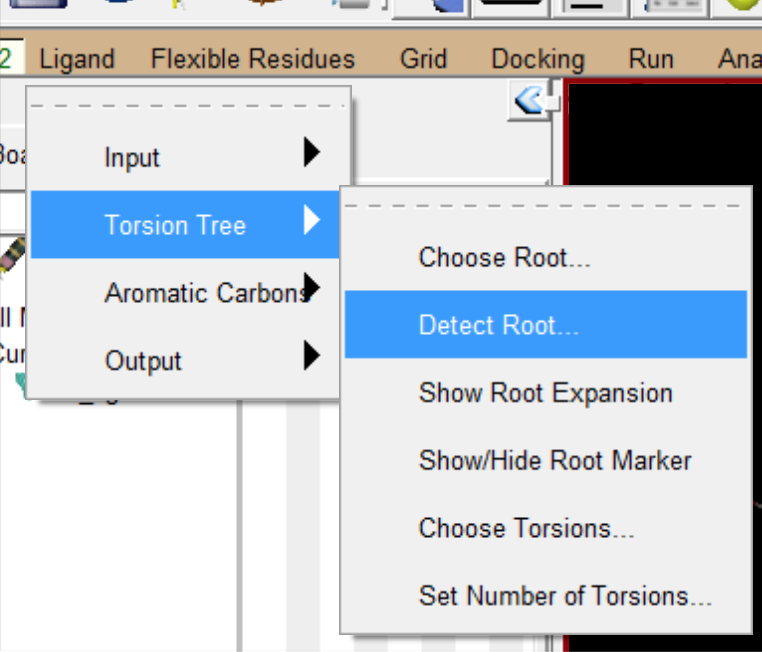
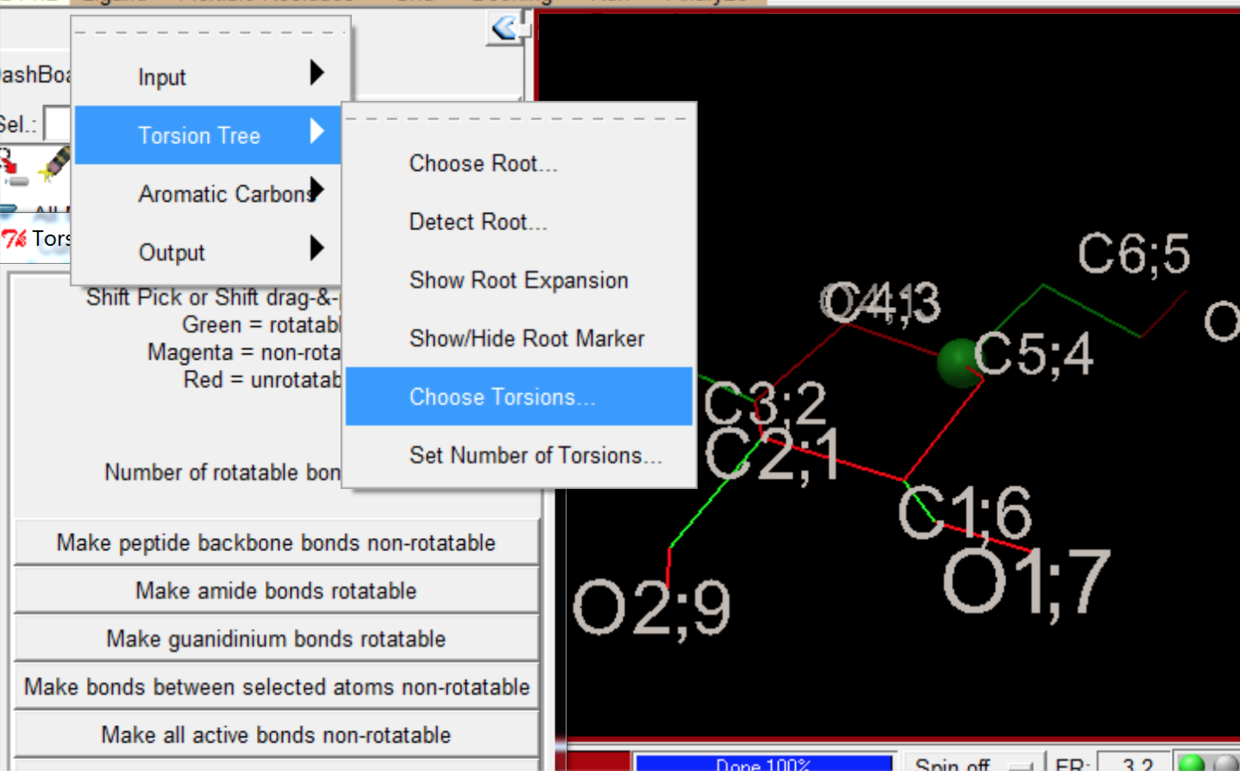
- 找出可旋转键
- 导出为pdbqt 文件,配体就设置完成了。
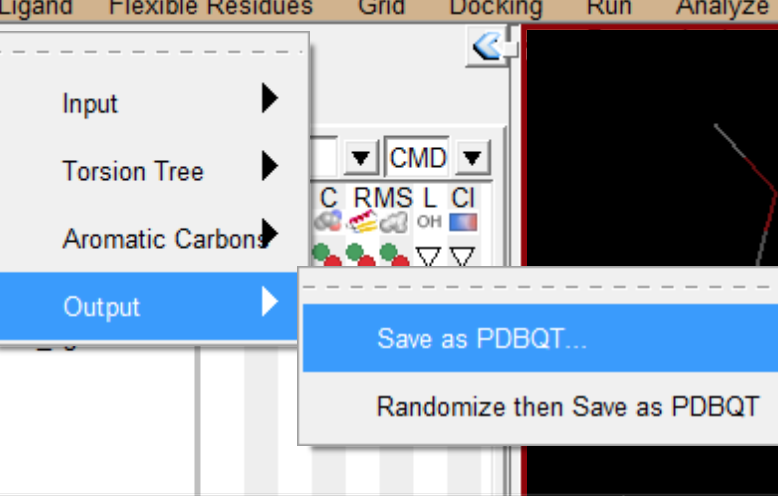
- 同配体一样,预处理(氢、电荷)
- 将蛋白质设定为对接蛋白质
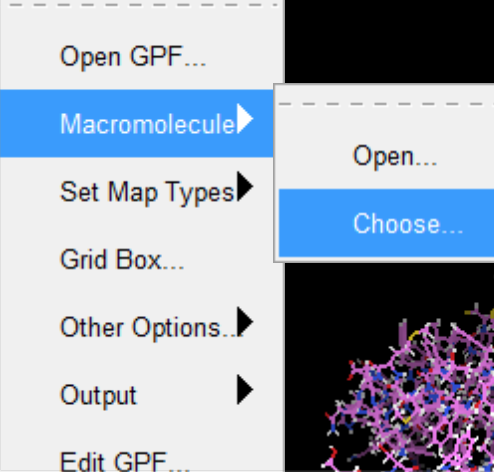
- 设置对接参数

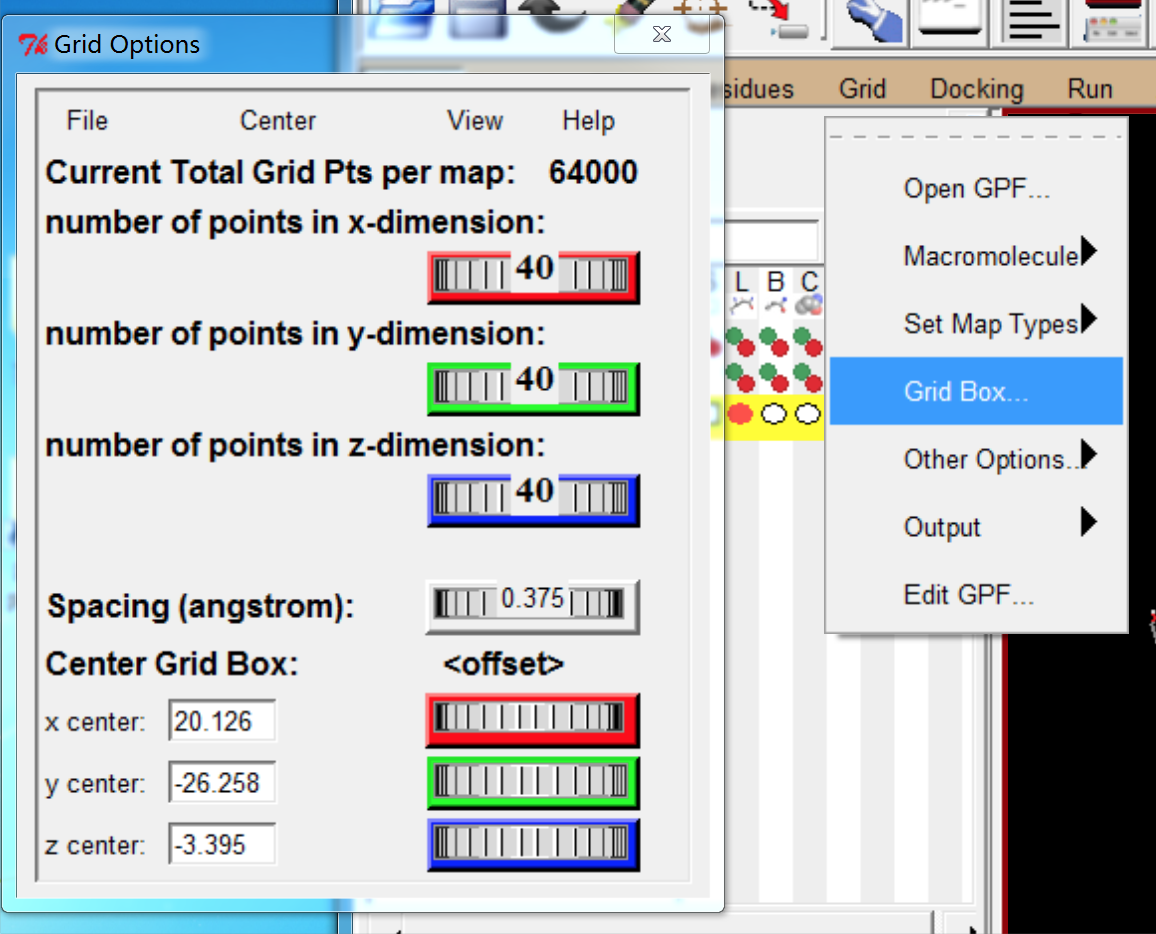
- box 的设定可以参考该蛋白质和其他已发表文章中的小分子对接的位置。

- 导出gpf 文件。
- 使用autodock4
打开终端,进入导出的gpf 文件目录下。
$ autogrid4 -p test.gpf -l test.glg
- 继续回到autodock 界面。在docking 栏目下选择rigid 文件,即之前保存的蛋白的pdbqt 文件。
- 接着在ligend 栏目里选择open,打开配体的pdbqt文件。并接受默认参数设定。
- 一共有第三个算得快,但第一个算得更精准(耗时也长一些)。
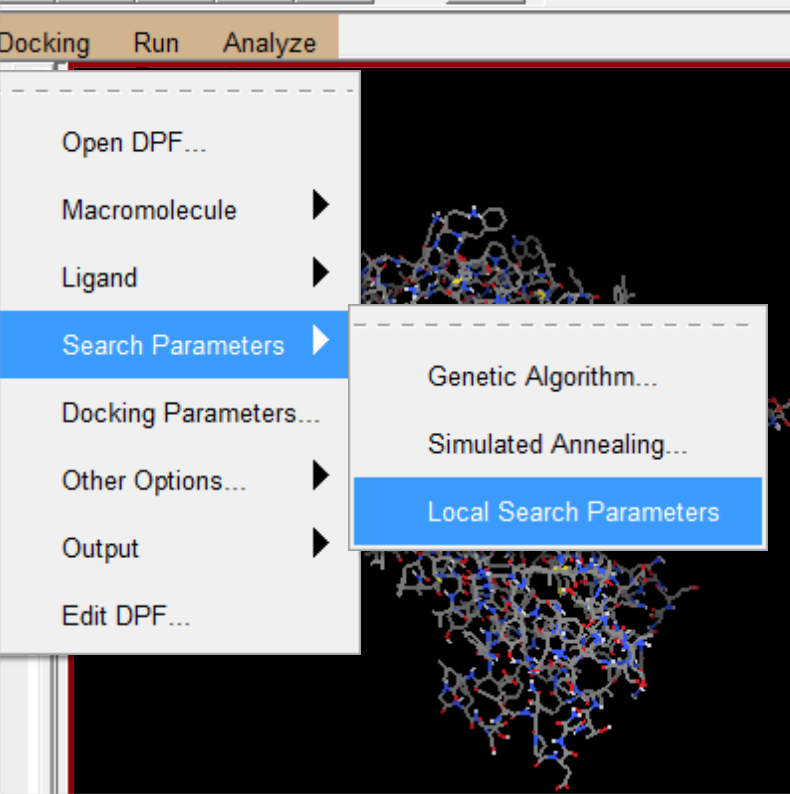
- 再选择docking parameter,接受默认选项。
- 输出docking 文件
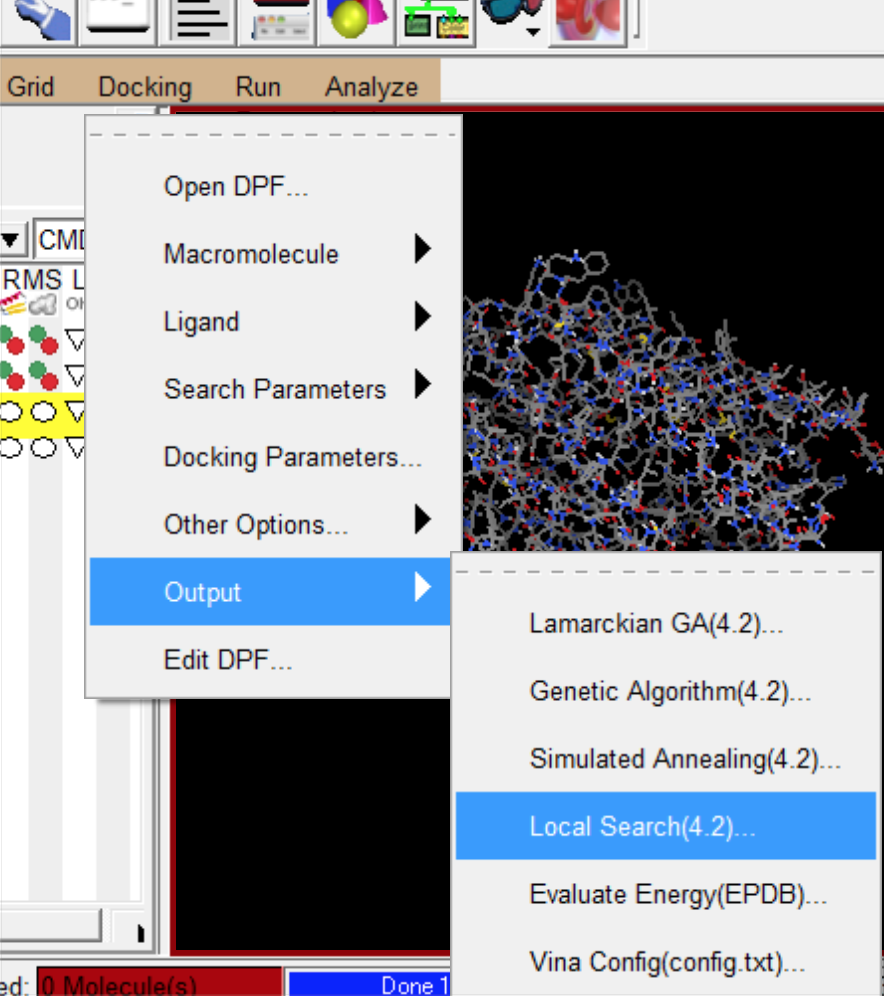
- 再回到终端,开始对接
$ autodock4 -p test.dpf -l test.dlg
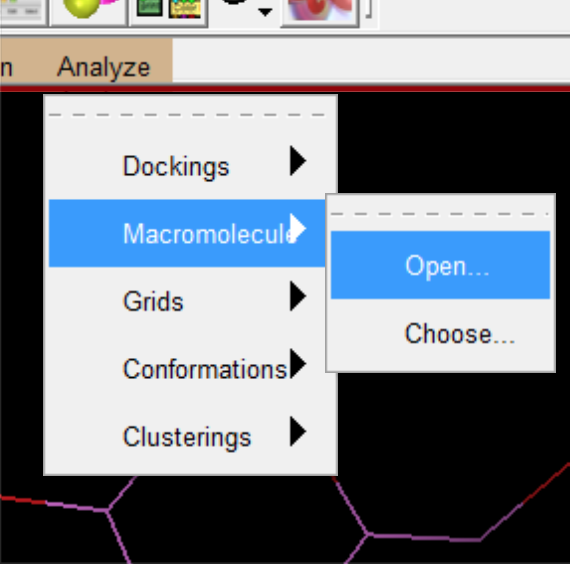
- 删除之前载入的内容,导入对接结果(dlg文件),和蛋白质文件。
导入后的初始内容,为对接前的结果。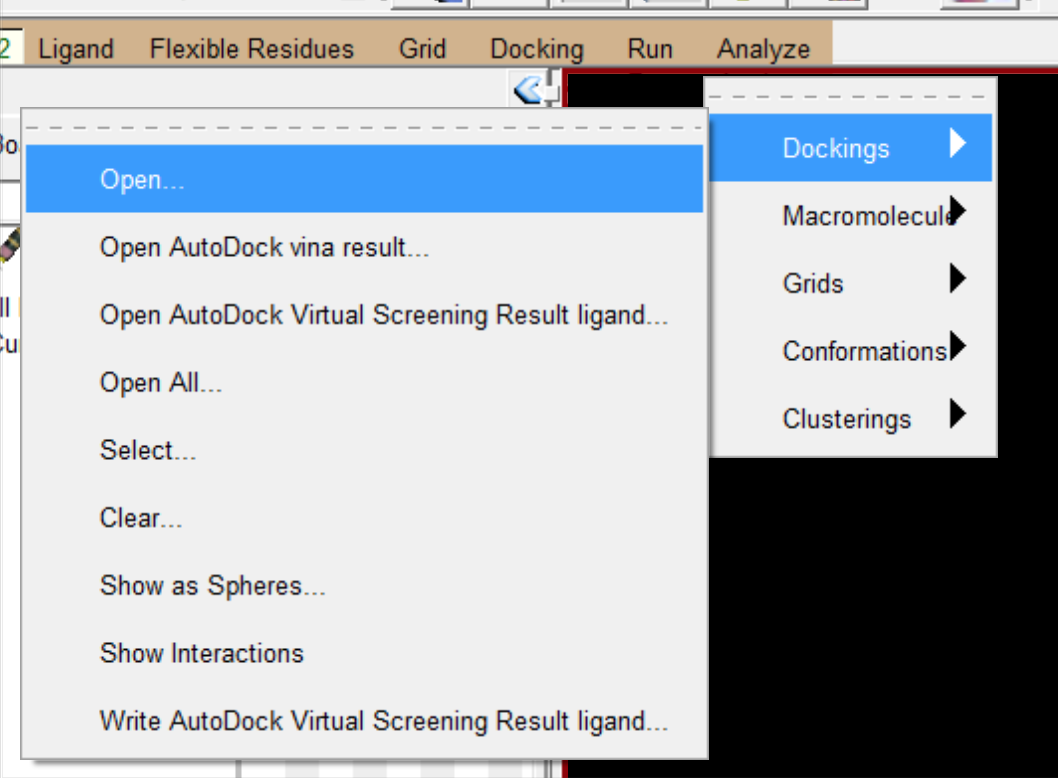
- 这个工具条可以将结果根据能量由低到高显示出来。
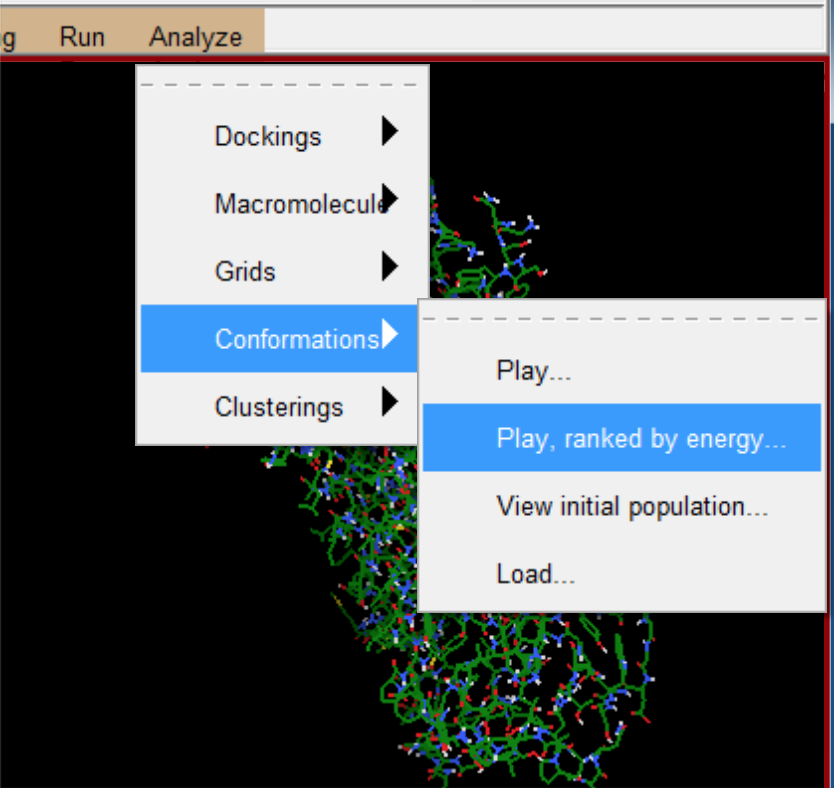
- 点击play 可以看到前50个对接的结果。(按顺序第一个则比对最好,排名最高)
- 可以看到对接结果能量都是小于0的
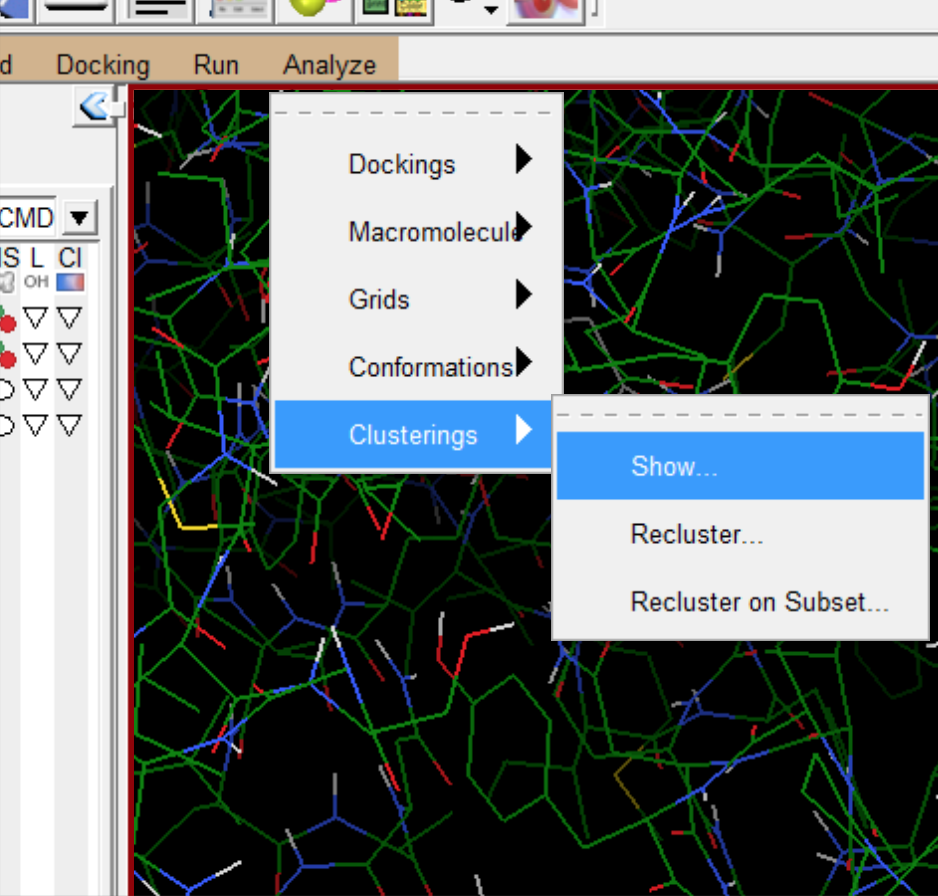
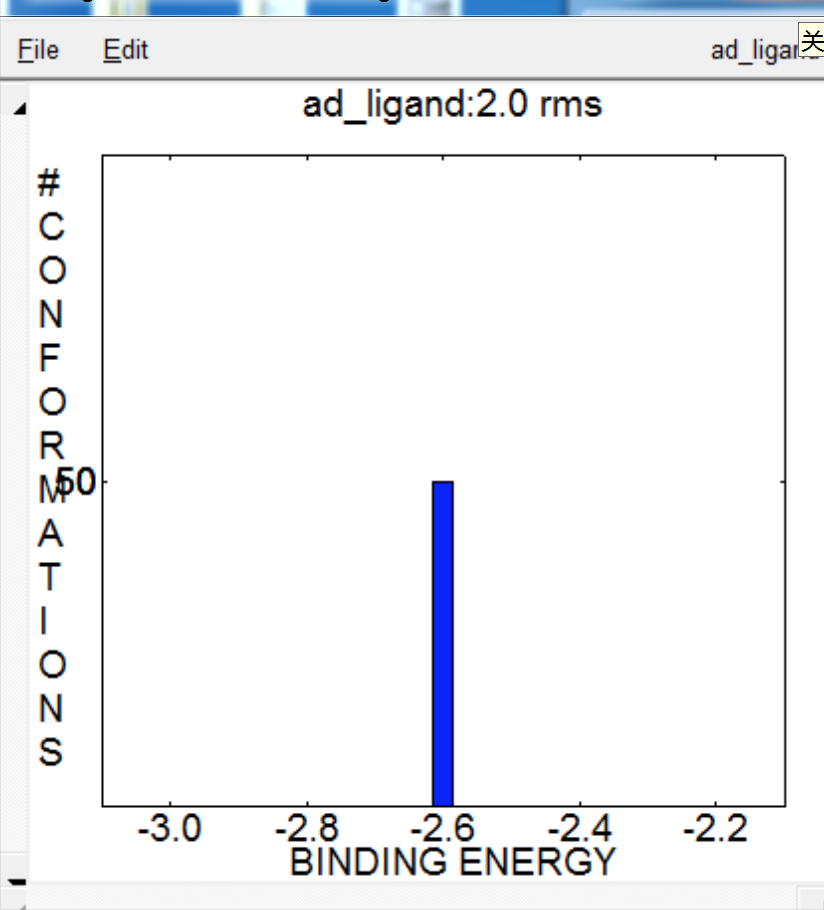
- 查看小分子和蛋白相互作用的细节。可以看到蛋白质与小分子产生氢键的位置。
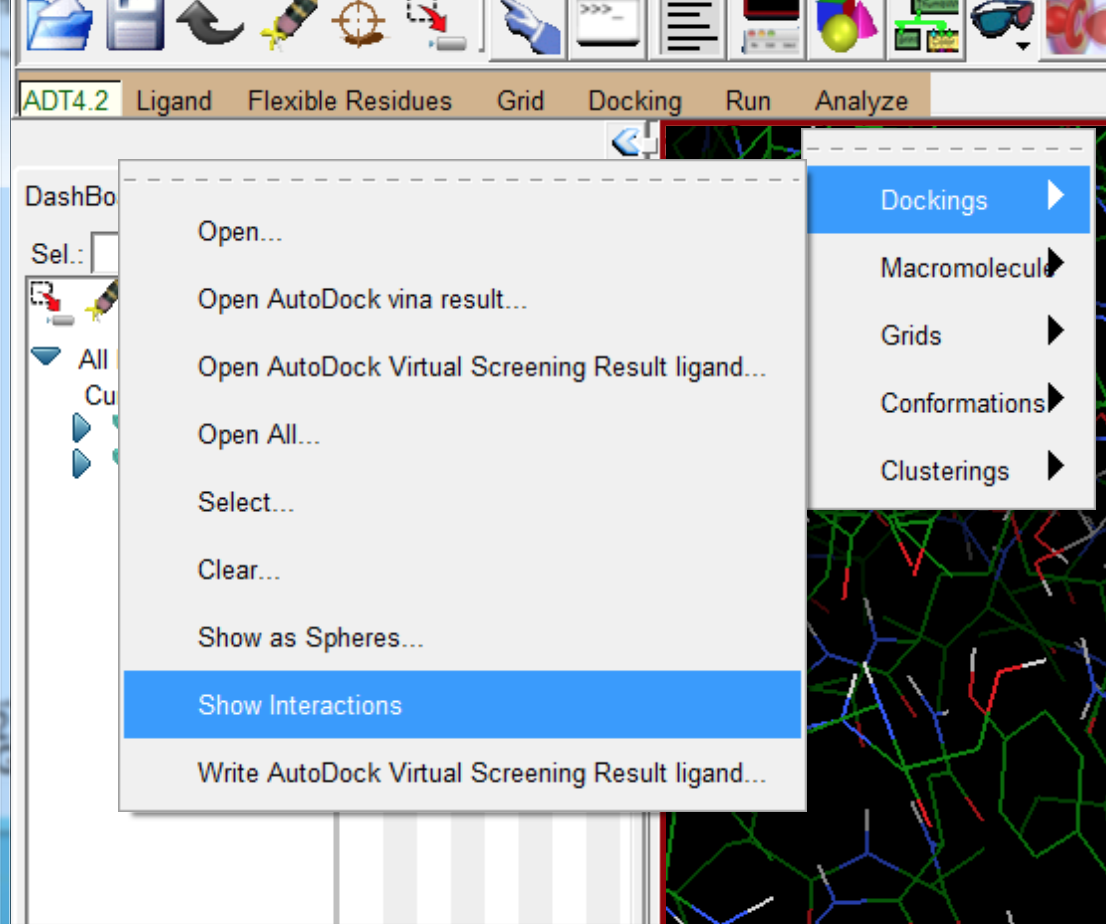
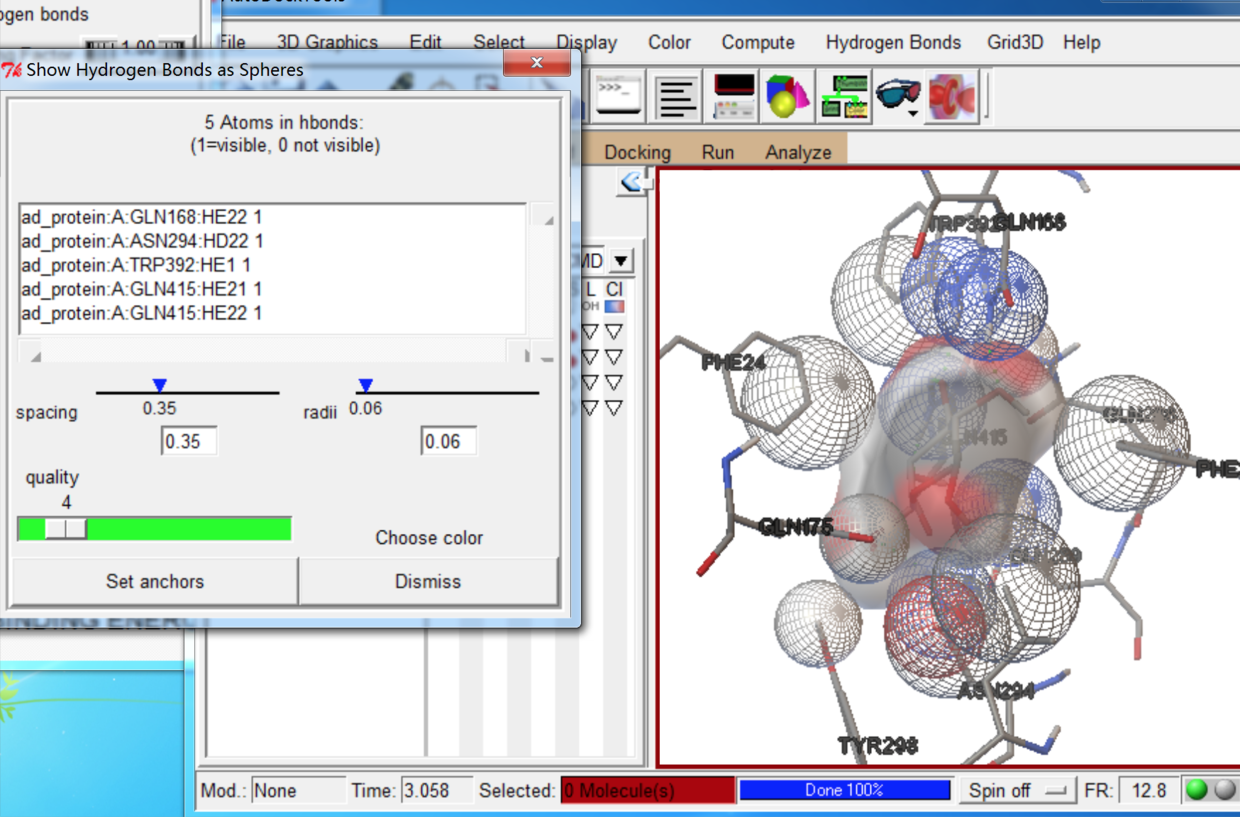
- 只保留排名最高的比对结果。并保存结果。
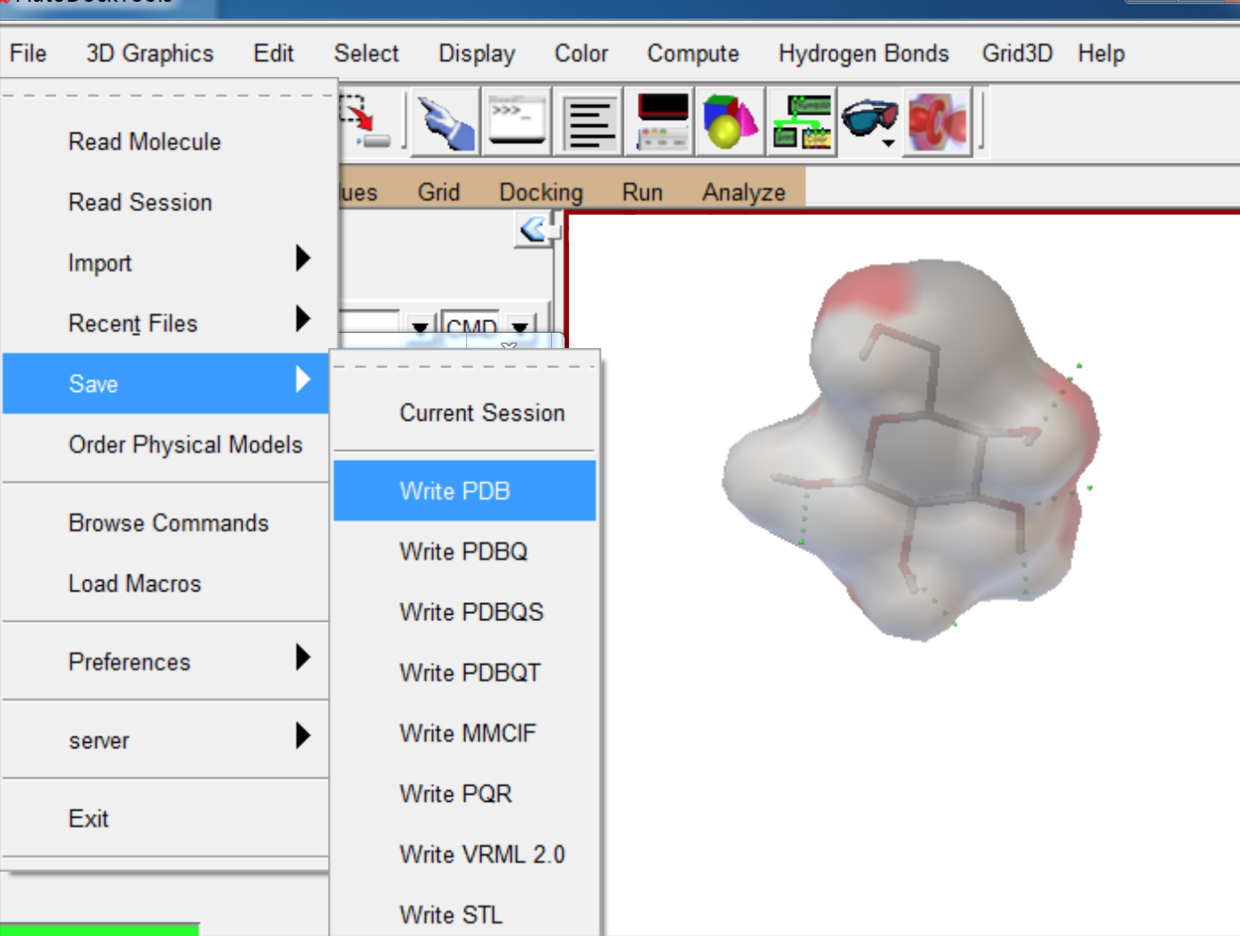
- 将文件信息合并到原蛋白pdb文件,形成小分子-蛋白复合物的pdb 文件。接着可以用vmd 等软件做进一步处理。

若有收获,就点个赞吧
0 人点赞
