构建系统发生树的意义
- 对于一个未知的基因或蛋白质序列,确定其亲缘关系最近的物种。
- 预测一个新发现的基因或蛋白质功能。
- 有助于预测一个分子功能的走势。
- 追溯一个基因的起源
什么是系统发生树
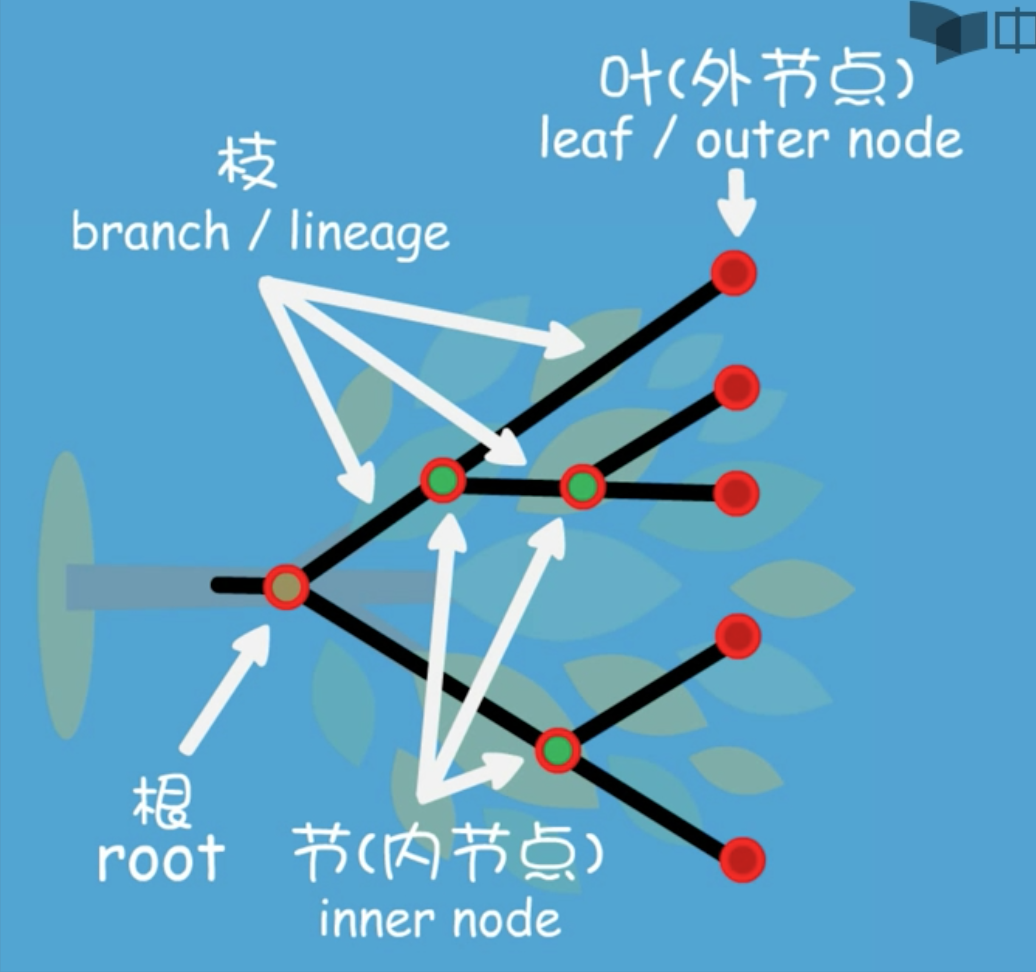
现有生物都在叶节点上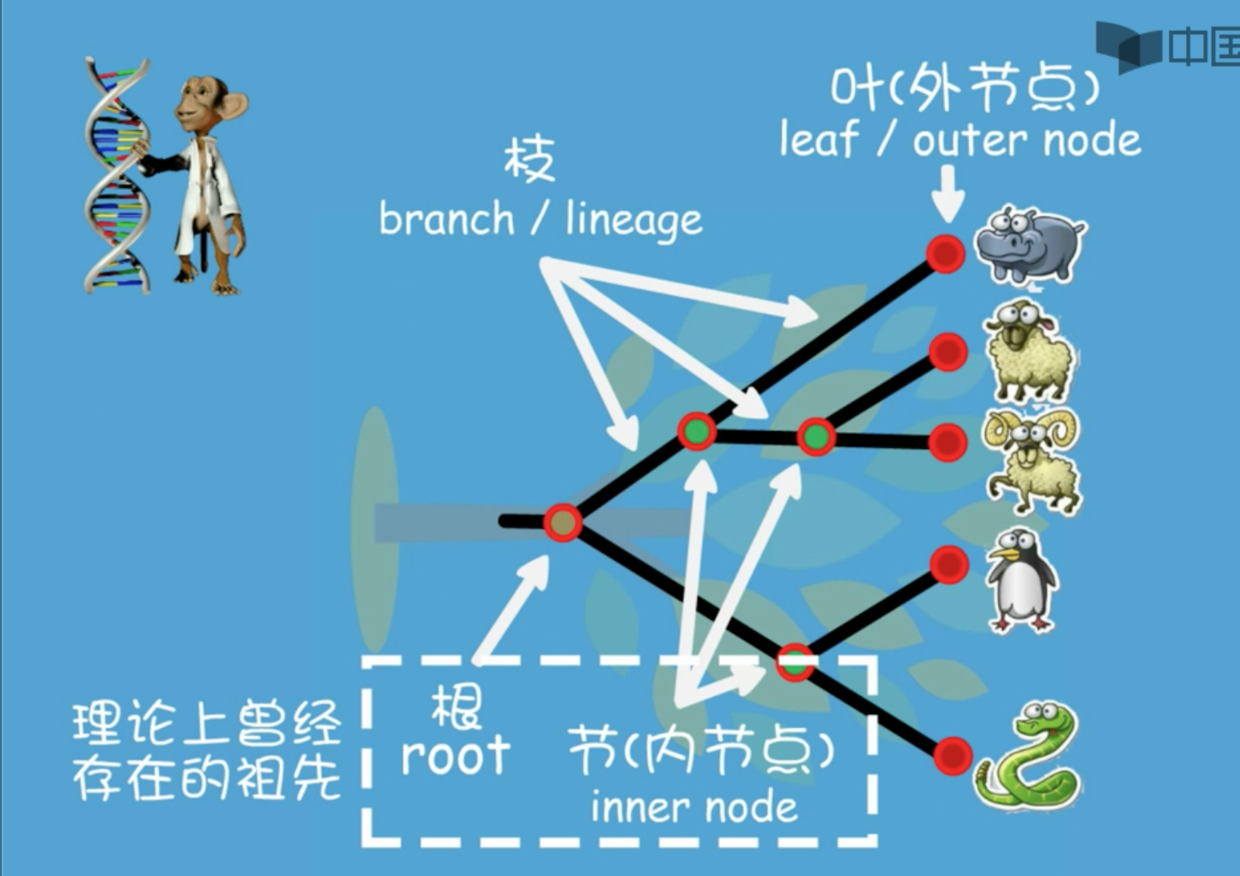
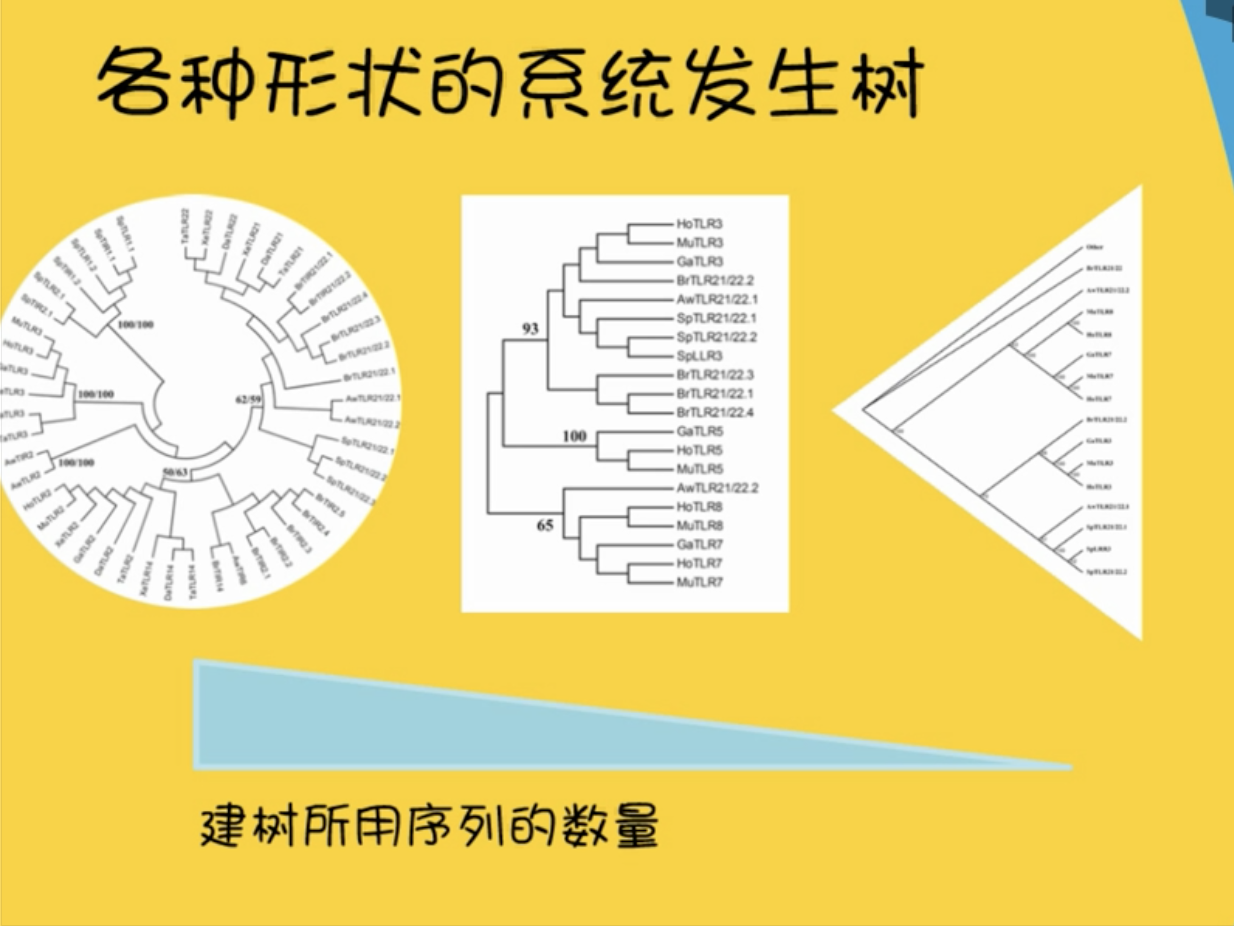
各种各样的系统发生树
系统发生树的特性
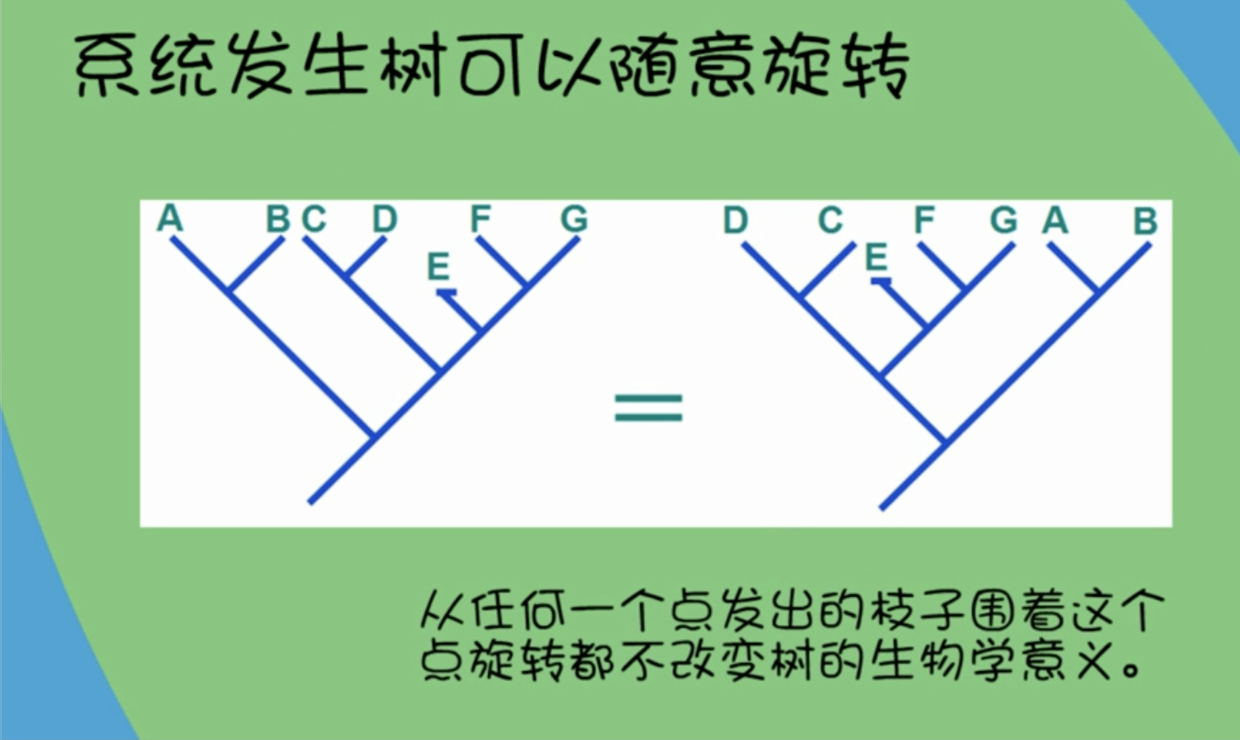
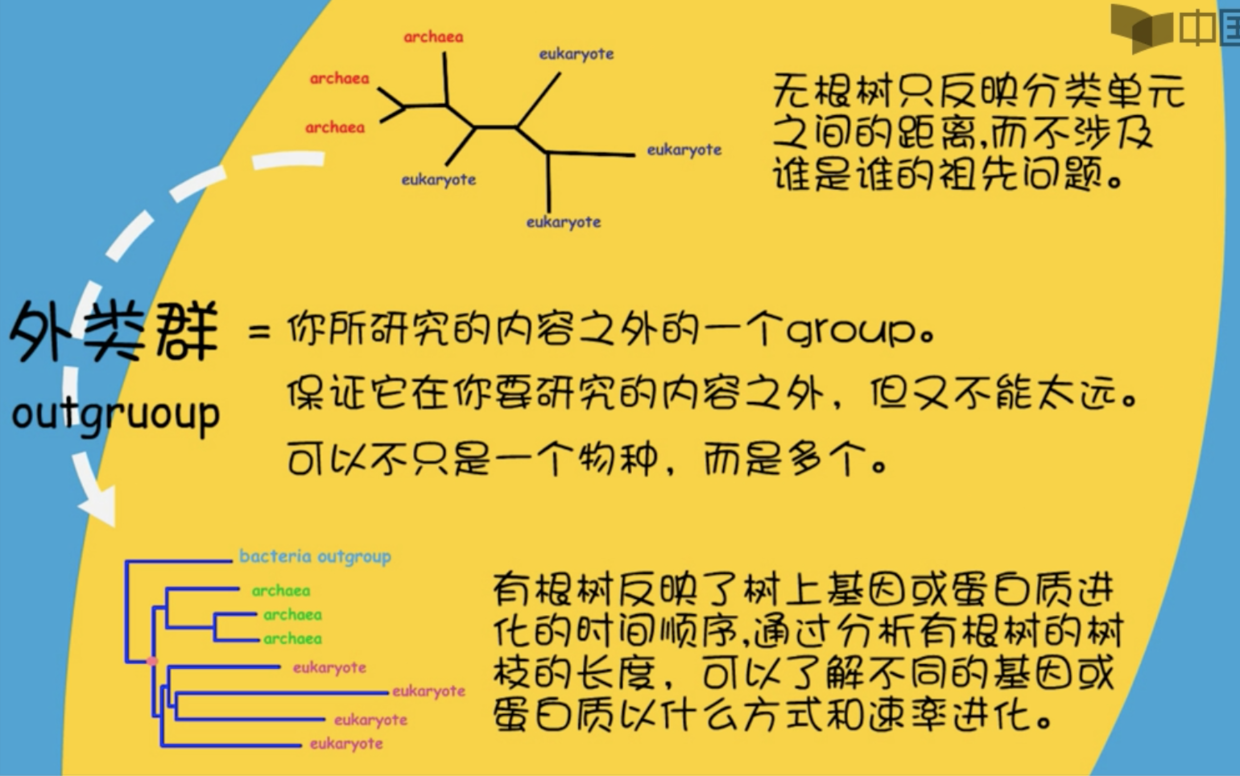
- 对于系统发生树来说,根的位置是主观的
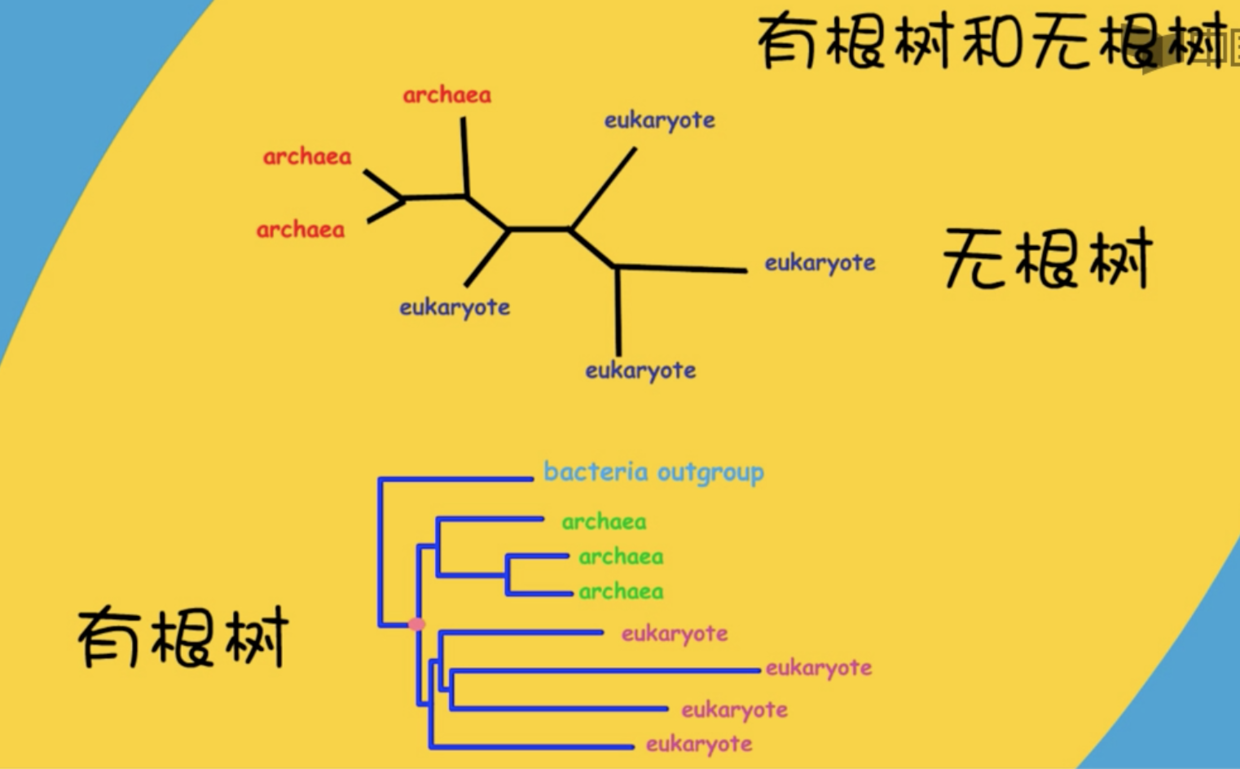
- 做有根树,需要指定外类群
因为我们知道外类群和现有的研究对象一定不是一个物种,因此分叉出来的那个节点,就一定是根。
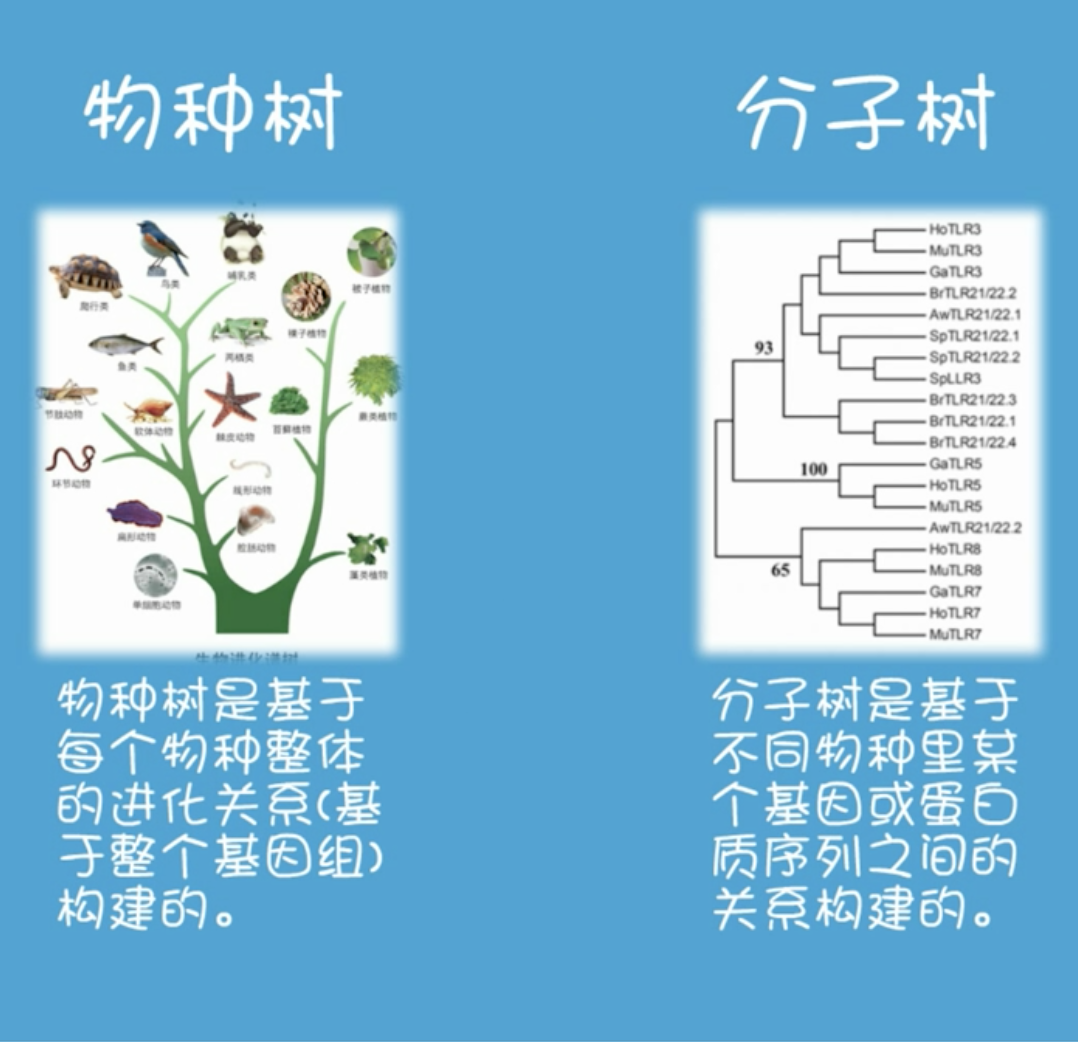
分子树与物种树的差别
构建系统发生树
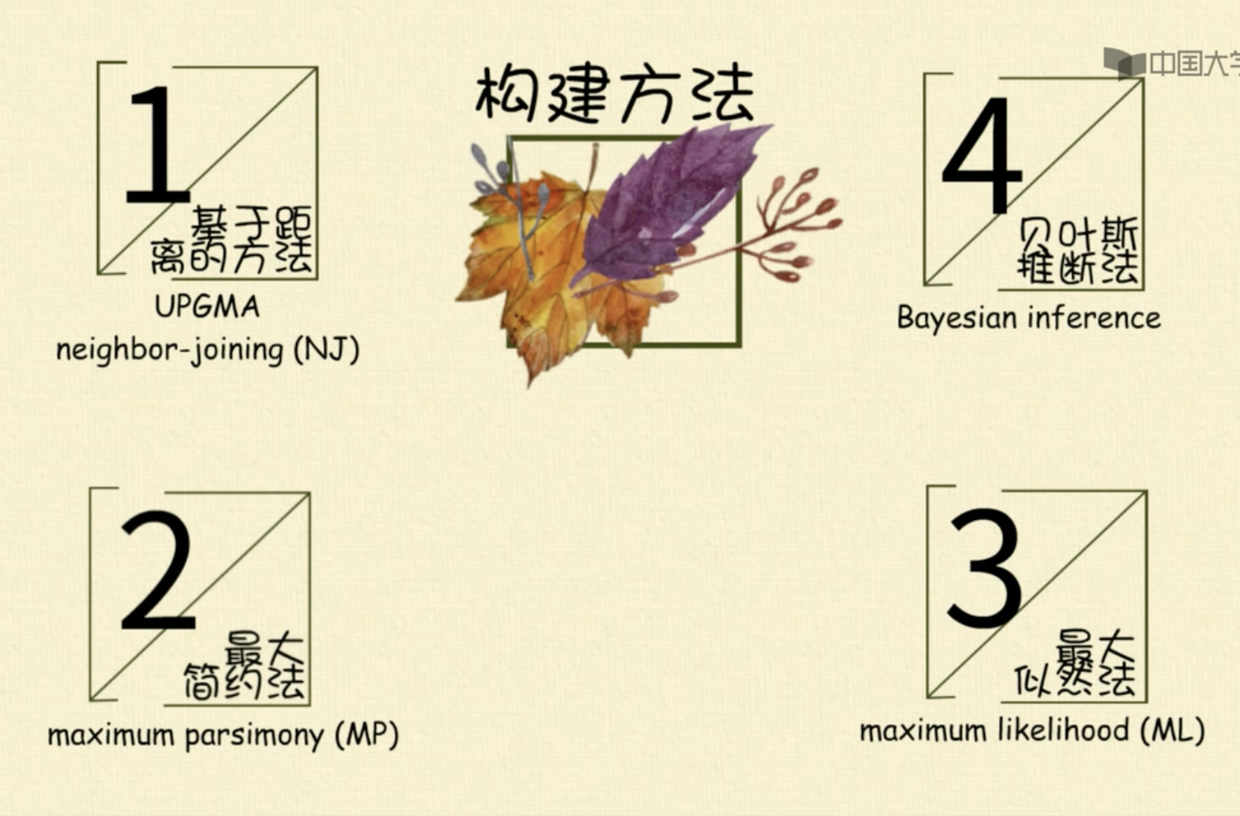
按速度来看:基于距离的方法> 最大简约法> 最大似然法> 贝叶斯推断法
但相应来看,速度越快,其准确度也越低。
序列的选择

看DNA 序列一致度是否大于70 %。
构建系统发生树的软件

UPGMA 法

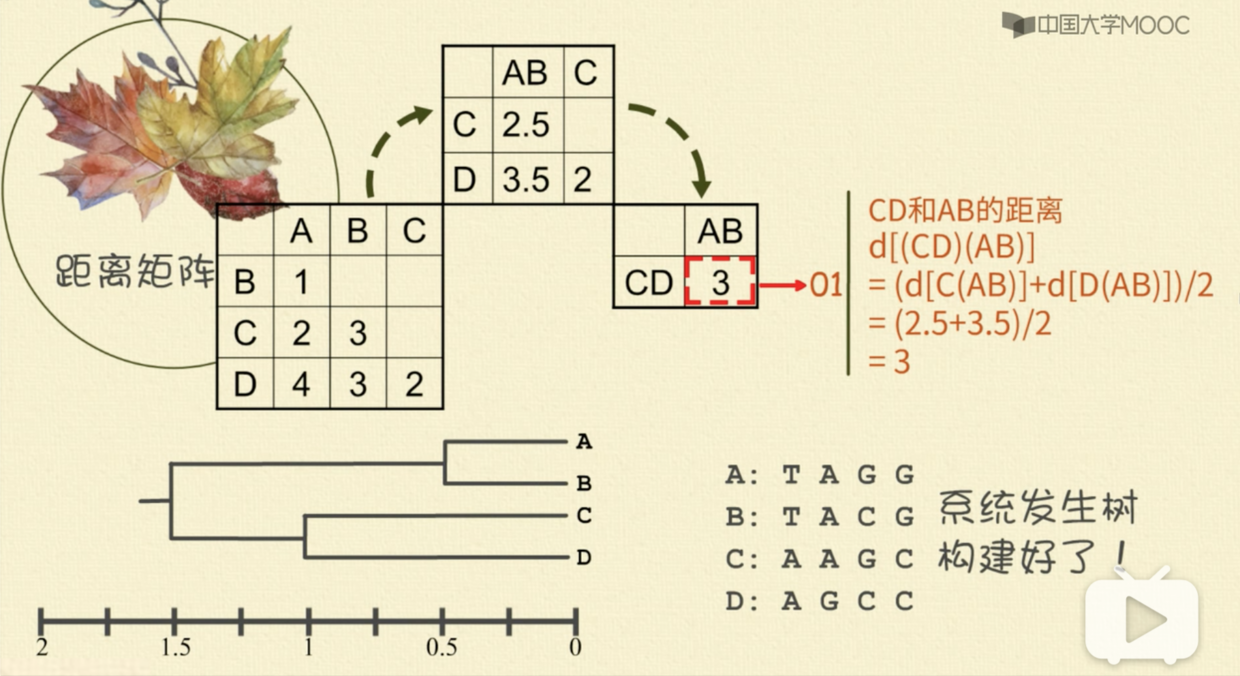
- 树上枝条的长短,直接反映了它们与共同祖先的距离。
使用mega 构建系统发生树
- 通过mega 官网,就可以直接下载这款软件了。(我使用的是mac 的图形化版本)
- 首先我们需要准备一份fasta 格式的文件,里面包含了需要进行比对的序列的全部信息。
- 导入序列后,选择
align就可以进行比对。 - 进入后再在
alignment的操作栏中选定align by clustalW,就可以使用该方法进行多序列比对了。 - 将比对后的比对文件导出为
MEGA format - 将新的比对文件使用mega 打开(可以直接拖拽至窗口)
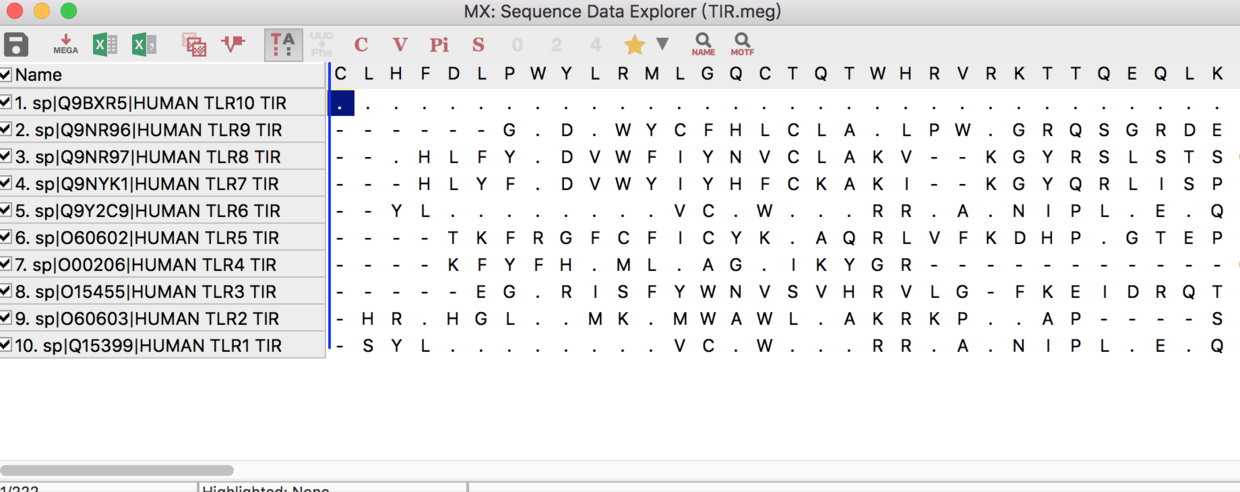
其中 TA 选项可以将相同的比对转换为-,不同的再特别标记出来。
C 标记保守序列,V 标记不保守序列。
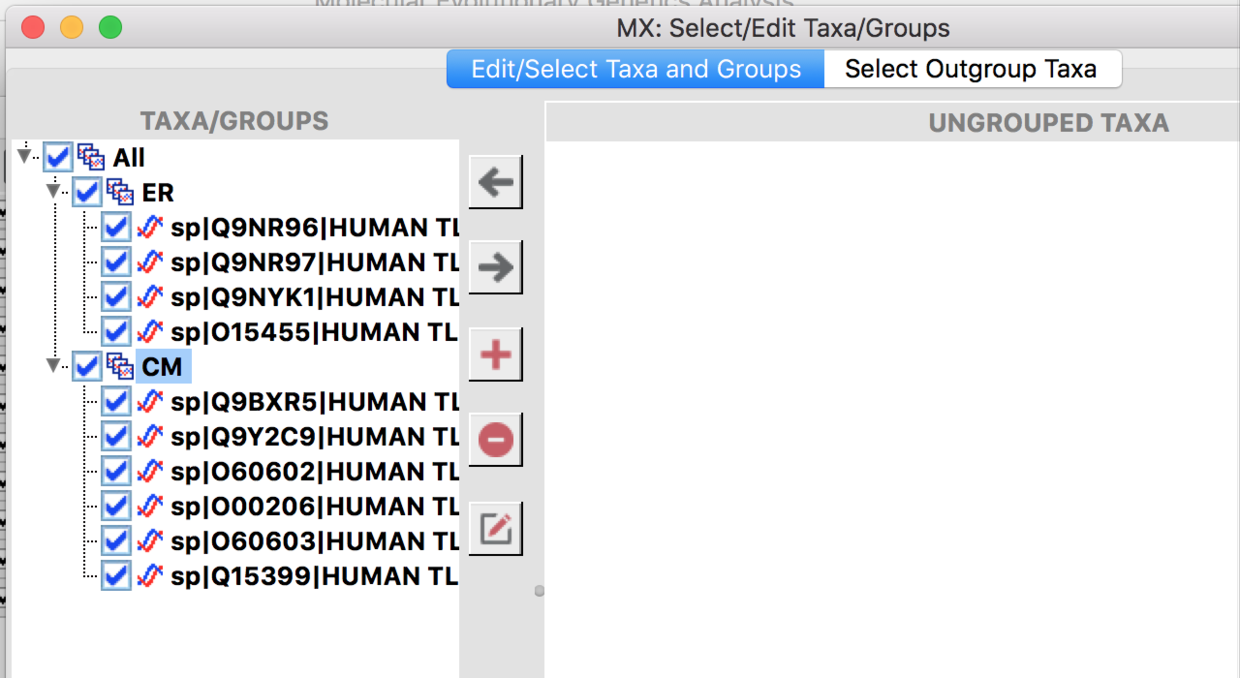
还可以为它们进行分组。
一般这里会将树名简短一些,方便后期显示。
- 使用phylogeny 进行建树,使用默认参数即可。
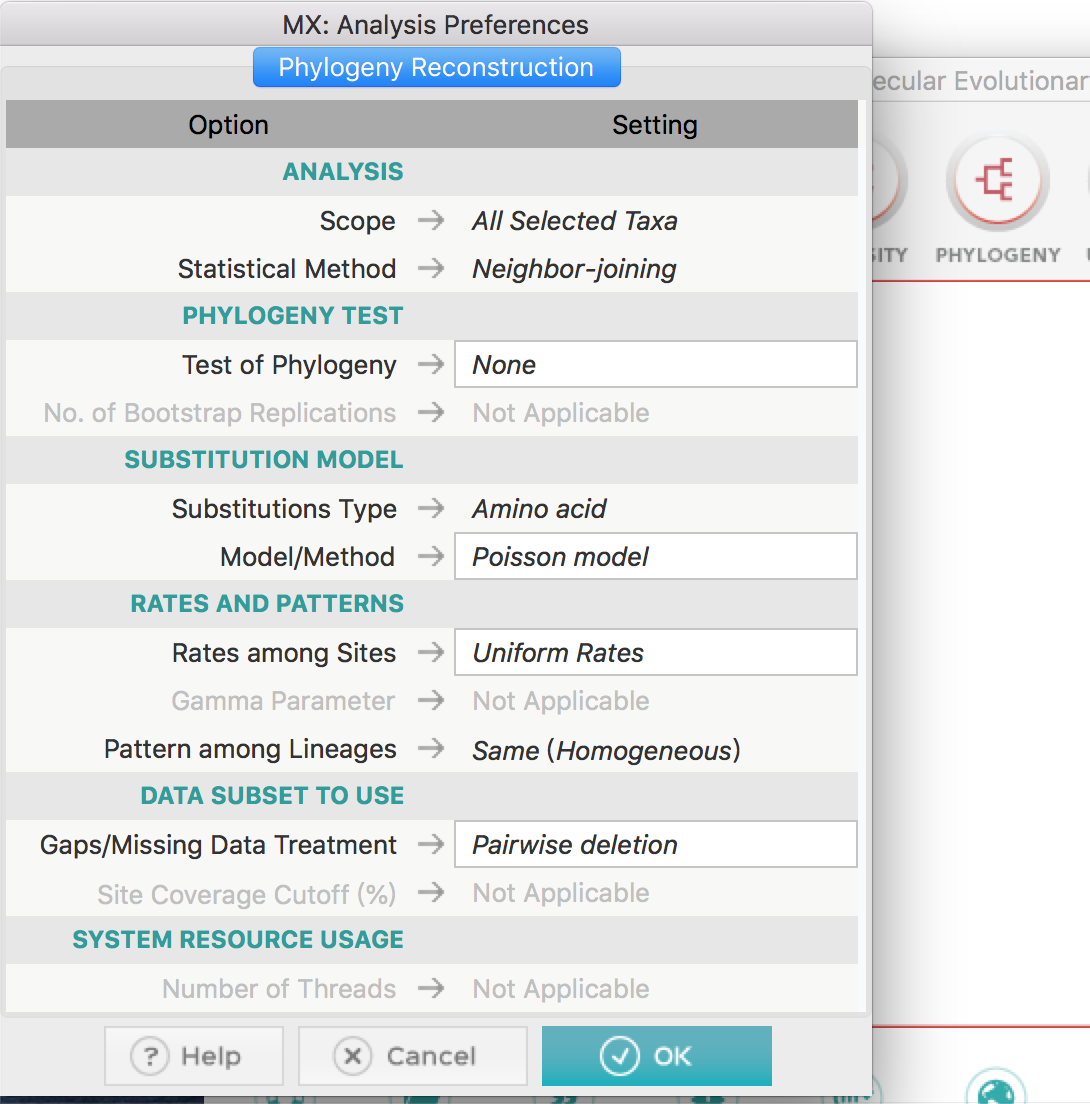
test of phylogeny:建树的检验方法,默认不进行检验。
我们可以修改为Bootstrap method,通常为100的倍数,设定500。
通过该检验方法得到的系统发生树的每个节点都会标记一个数字,它代表了指定次数次所得到的系统发生树都百分之多少都有该节点,一般70%才可信。
Substitutions Model:选择计算遗传距离时使用的计算模型。理论上应该选择各种模型,根据各组结果,进行选定。但一般实际操作直接选用p-distance。
Gaps/Missing Data Treatment:大多数建树方法会要求删除有空位的链。根据统计方法,比如N-J方法, 选择partial deletion即可,删除程度定位50%即可。
- 选择
compute - 一共有两个树
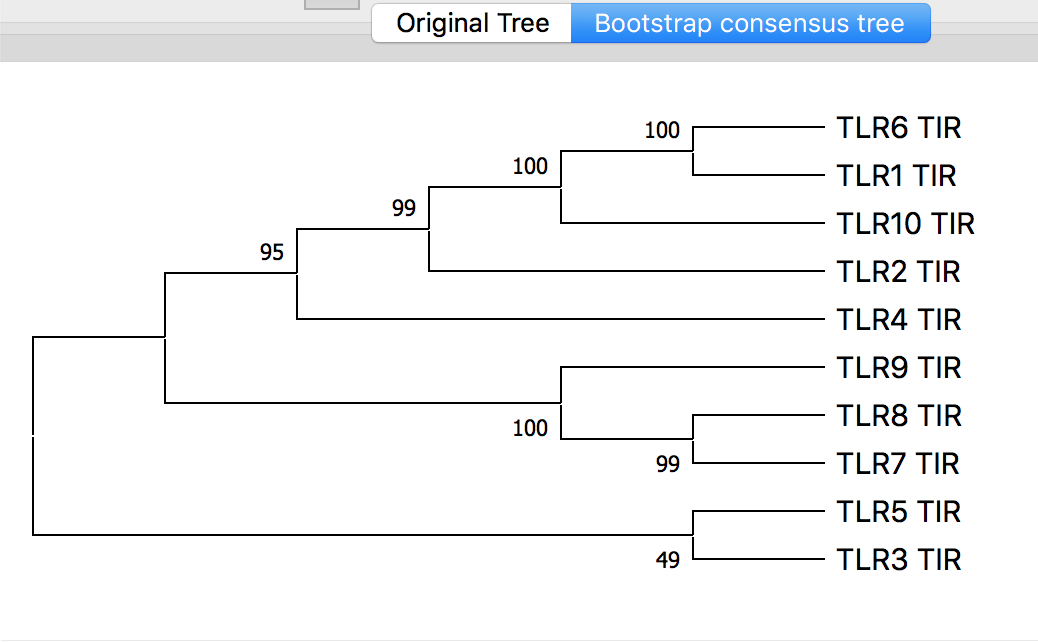
- 树中节点的数字表示,经过步长检验,有多少树包含该节点。
- 原始树为步长检验的五百颗树中的一个。未经过合并,因此树的长短可以精确代表
遗传距离。
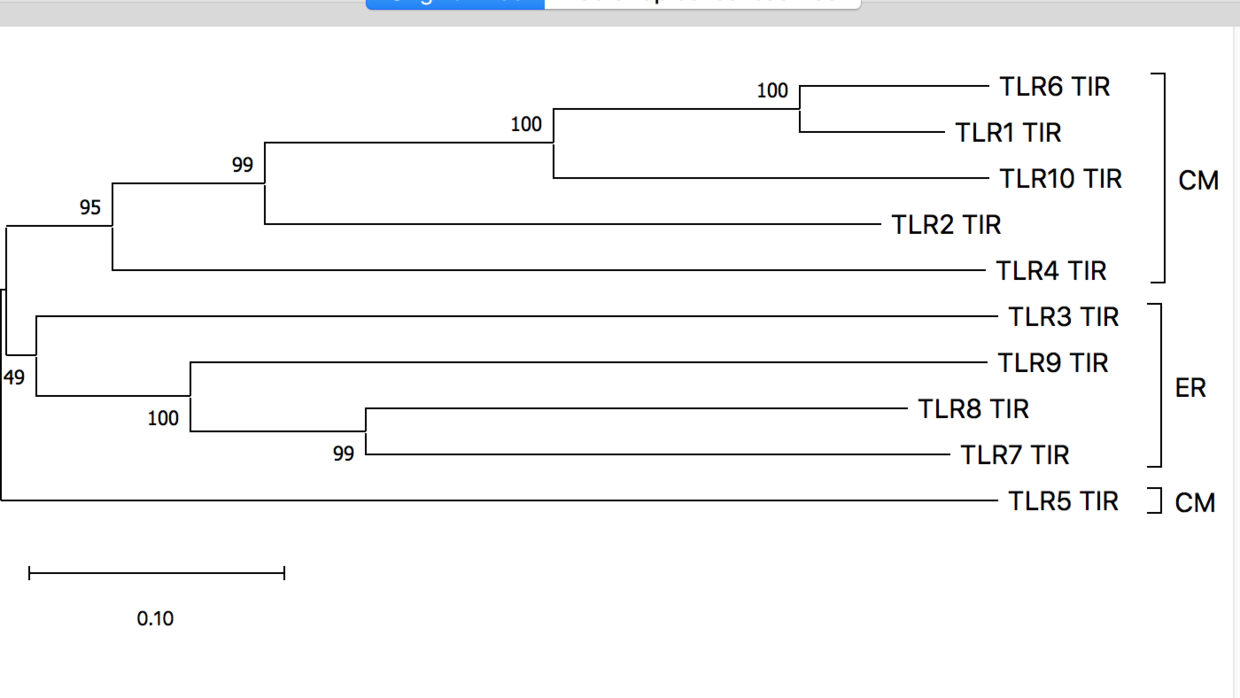
- 此外还可以设定发生树的图形,可以转变树枝或者选择自定义的树干。

若有收获,就点个赞吧
0 人点赞
