Finding a Most Likely Common Ancestor
In “Counting Point Mutations”, we calculated the minimum number of symbol mismatches between two strings of equal length to model the problem of finding the minimum number of point mutations occurring on the evolutionary path between two homologous strands of DNA. If we instead have several homologous strands that we wish to analyze simultaneously, then the natural problem is to find an average-case strand to represent the most likely common ancestor of the given strands.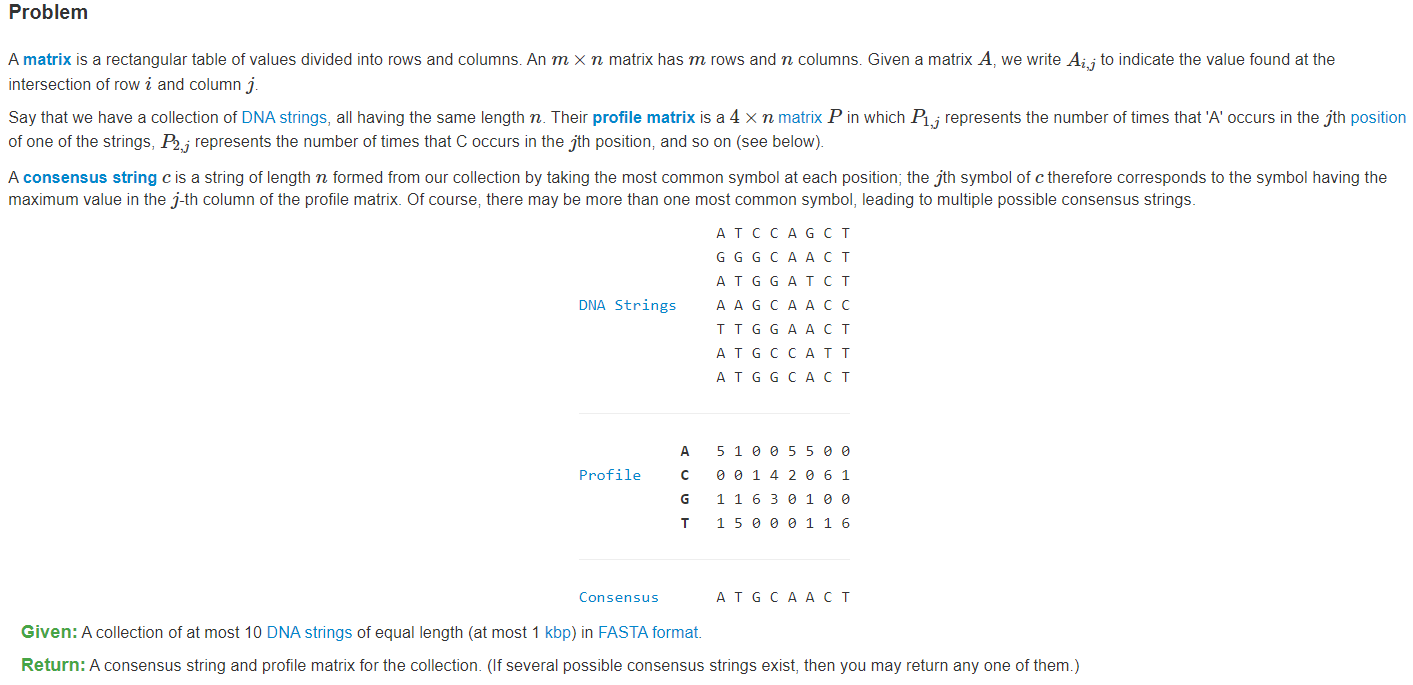
Sample Dataset
>Rosalind_1ATCCAGCT>Rosalind_2GGGCAACT>Rosalind_3ATGGATCT>Rosalind_4AAGCAACC>Rosalind_5TTGGAACT>Rosalind_6ATGCCATT>Rosalind_7ATGGCACT
Sample Output
ATGCAACTA: 5 1 0 0 5 5 0 0C: 0 0 1 4 2 0 6 1G: 1 1 6 3 0 1 0 0T: 1 5 0 0 0 1 1 6
Solution
本题不得不涉及文件操作,也是生物信息学上必须掌握读取 fasta 格式文件的方式。以 > 作为一条序列的开始,那么也是一条序列的结束。最难处理的就是第一条序列和第二条序列。
- 第一条序列:因为以
>作为序列终止,因此第一次遇到>需要判断seq是否为空,即第一条! - 最后一条序列:由于没有后继的
>作为终止,因此最后需要进一步筛选放入。
还有程序要尽量处理一些没用的空白符问题。
然后就是序列普问题,使用一个字典,一个键对应 len(seqs[0]) 长度的列表,然后遍历序列,分别统计每条序列对应位置上的碱基出现次数。最后再求出每个位置最大的就是最保守的序列。
from typing import Listclass Solution:def readFasta(self, fileName: str) -> List[str]:seqs = []with open(fileName) as f:seq = []for line in f:line = line.strip() # withespaceif line.startswith('>'): # IDif seq: # last sequence finishedseqs.append(''.join(seq))seq = []elif line: # basesseq.append(line.strip())# last sequenceif seq:seqs.append(''.join(seq))print(seqs)return seqsdef calculateProfile(self) -> None:seqs = self.readFasta('./DNA.fasta')if not seqs or not seqs[0]: returnk = len(seqs[0])d = {base: [0] * k for base in 'AGCT'}for seq in seqs:for idx, base in enumerate(seq):d[base][idx] += 1print(''.join((max(d, key=lambda x: d[x][i]) for i in range(k))))for base in 'ACGT':print(f'{base}:', ' '.join(map(str, d[base])))Solution().calculateProfile()'''['ATCCAGCT', 'GGGCAACT', 'ATGGATCT', 'AAGCAACC', 'TTGGAACT', 'ATGCCATT', 'ATGGCACT']ATGCAACTA: 5 1 0 0 5 5 0 0C: 0 0 1 4 2 0 6 1G: 1 1 6 3 0 1 0 0T: 1 5 0 0 0 1 1'''

若有收获,就点个赞吧
0 人点赞
