Modeling Random Genomes
We already know that the genome is not just a random strand of nucleotides; recall from “Finding a Motif in DNA” that motifs recur commonly across individuals and species. If a DNA motif occurs in many different organisms, then chances are good that it serves an important function.
我们已经知道,基因组不仅是核苷酸的随机链;而且 回想一下“在DNA中找到模体”,主题在个人和物种中普遍存在。如果DNA基序出现在许多不同的生物中,则很有可能发挥其重要功能。
At the same time, if you form a long enough DNA string, then you should theoretically be able to locate every possible short substring in the string. And genomes are very long; the human genome contains about 3.2 billion base pairs. As a result, when analyzing an unknown piece of DNA, we should try to ensure that a motif does not occur out of random chance.
同时,如果形成足够长的DNA字符串,那么从理论上讲,您应该能够找到字符串中所有可能的短子字符串。同时基因组非常长; 人类基因组包含约32亿个碱基对。所以,当分析未知的DNA片段时,我们应尝试确保 motif 不是偶然出现的。
To conclude whether motifs are random or not, we need to quantify the likelihood of finding a given motif randomly. If a motif occurs randomly with high probability, then how can we really compare two organisms to begin with? In other words, all very short DNA strings will appear randomly in a genome, and very few long strings will appear; what is the critical motif length at which we can throw out random chance and conclude that a motif appears in a genome for a reason?
为了确定 motif 是否随机出现,我们需要量化随机找到给定 motif 的可能性。如果一个 motif 很有可能是随机出现,那么我们如何才能真正比较两种生物呢?
In this problem, our first step toward understanding random occurrences of strings is to form a simple model for constructing genomes randomly. We will then apply this model to a somewhat simplified exercise: calculating the probability of a given motif occurring randomly at a fixed location in the genome.
Problem**
An array is a structure containing an ordered collection of objects (numbers, strings, other arrays, etc.). We let A[k] denote the k-th value in array A. You may like to think of an array as simply a matrix having only one row.
A random string is constructed so that the probability of choosing each subsequent symbol is based on a fixed underlying symbol frequency.
GC-content offers us natural symbol frequencies for constructing random DNA strings. If the GC-content is x, then we set the symbol frequencies of C and G equal to  and the symbol frequencies of A and T equal to
and the symbol frequencies of A and T equal to  . For example, if the GC-content is 40%, then as we construct the string, the next symbol is ‘G’/‘C’ with probability 0.2, and the next symbol is ‘A’/‘T’ with probability 0.3.
. For example, if the GC-content is 40%, then as we construct the string, the next symbol is ‘G’/‘C’ with probability 0.2, and the next symbol is ‘A’/‘T’ with probability 0.3.
In practice, many probabilities wind up being very small. In order to work with small probabilities, we may plug them into a function that “blows them up” for the sake of comparison. Specifically, the common logarithm of x (defined for x>0 and denoted log10(x)) is the exponent to which we must raise 10 to obtain x.
See Figure 1 for a graph of the common logarithm function y=log10(x). In this graph, we can see that the logarithm of x-values between 0 and 1 always winds up mapping to y-values between −∞ and 0: x-values near 0 have logarithms close to −∞, and x-values close to 1 have logarithms close to 0. Thus, we will select the common logarithm as our function to “blow up” small probability values for comparison.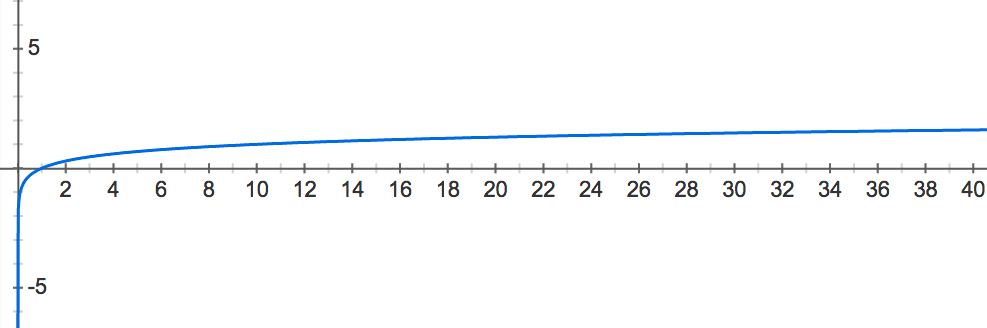
Figure 1. The graph of the common logarithm function of x. For a given x-value, the corresponding y-value is the exponent to which we must raise 10 to obtain x. Note that x-values between 0 and 1 get mapped to y-values between -infinity and 0.
Given: A DNA string s of length at most 100 bp and an array A containing at most 20 numbers between 0 and 1.
Return: An array B having the same length as A in which B[k] represents the common logarithm of the probability that a random string constructed with the GC-content found in A[k] will match s exactly.
Sample Dataset
ACGATACAA
0.129 0.287 0.423 0.476 0.641 0.742 0.783
Sample Output
-5.737 -5.217 -5.263 -5.360 -5.958 -6.628 -7.009
Hint
One property of the logarithm function is that for any positive numbers x and y, log10(x⋅y)=log10(x)+log10(y).
Solution
其实本题说的很有用,尤其是对数据分析。
怎么要随机化构建 DNA 序列呢?题目给出 就是 GC 含量,根据题意  ,同时
,同时  。题目要求通过每个不同
。题目要求通过每个不同  (
( GC 含量),判断是随机构成  串的概率是多少。因此将
串的概率是多少。因此将  每位出现概率累乘即可
每位出现概率累乘即可  。
。
为什么要用对数?假设现在不用对数,我们进行累乘,最终答案如下所示。
1.8306617184691366e-066.066146693326933e-065.455141499014318e-064.360523376795354e-061.1012117052575903e-062.353206236231256e-079.789797943792126e-08
可见上面都是高精度浮点数,实际比较起来非常小,但是使用 y = l0g10(x) 目的就是将不同值之间的差距放大,这样对比比较明显。(相反也有缩小,如基因表达差异分析中使用的  即是如此)
即是如此)
import mathdata = """ACGATACAA0.129 0.287 0.423 0.476 0.641 0.742 0.783"""seq, arr = filter(bool, data.splitlines())arr = list(map(float, arr.split()))for p in arr:d = {'G' : p / 2, 'C': p / 2, 'T': (1 - p) / 2, 'A': (1 - p) / 2}print(f'{sum(math.log10(d[base]) for base in seq):.3f}', end=' ')

若有收获,就点个赞吧
0 人点赞
