- fastqc
- 1 Basic Statistics
- Summary
- Warning
- Failure
- Common reasons for warnings
- 警告
- 失败
- 警告的常见原因
- 2 Per Base Sequence Quality
- Summary
- 摘要
- Warning
- 警告
- Failure
- 失败
- Common reasons for warnings
- 警告的常见原因
- 3 Per Sequence Quality Scores
- Summary
- 摘要
- Warning
- 警告
- Failure
- 失败
- Common reasons for warnings
- 警告的常见原因
- 4 Per Base Sequence Content
- Summary
- 摘要
- Warning
- 警告
- Failure
- 失败
- Common reasons for warnings
- 发出警告的常见原因
- 5 Per Sequence GC Content
- Summary
- 摘要
- Warning
- 警告
- Failure
- 失败
- Common reasons for warnings
- 警告的常见原因
- 6 Per Base N Content
- Summary
- Warning
- 警告
- Failure
- 失败
- Common reasons for warnings
- 警告的常见原因
- 7 Sequence Length Distribution
- Summary
- 摘要
- Warning
- 警告
- Failure
- 失败
- Common reasons for warnings
- 警告的常见原因
- 8 Duplicate Sequences
- Summary
- 摘要
- Warning
- 警告
- Failure
- 失败
- Common reasons for warnings
- 出现警告的常见原因
- 9 Overrepresented Sequences
- Summary
- 摘要
- Warning
- 警告
- Failure
- 失败
- Common reasons for warnings
- 警告的常见原因
- 10 Adapter Content
- Summary
- 摘要
- Warning
- Failure
- Common reasons for warnings
- 警告
- 失败
- 警告的常见原因
- 11 Kmer Content
- Summary
- Warning
- Failure
- Common reasons for warnings
- 警告
- 失败
- 警告的常见原因
- 12 Per Tile Sequence Quality
- Summary
- 摘要
- Warning
- Failure
- Common reasons for warnings
- 警告
- 失败
- 警告的常见原因
- multiqc
- FastQ Screen
fastqc
| Function | A quality control tool for high throughput sequence data. |
|---|---|
| Language | Java |
| Requirements | A suitable Java Runtime Environmen The Picard BAM/SAM Libraries (included in download) |
| Code Maturity | Stable. Mature code, but feedback is appreciated. |
| Code Released | Yes, under GPL v3 or later. |
| Initial Contact | Simon Andrews |
| FastQC aims to provide a simple way to do some quality control checks on raw sequence data coming from high throughput sequencing pipelines. It provides a modular set of analyses which you can use to give a quick impression of whether your data has any problems of which you should be aware before doing any further analysis. | FastQC旨在提供一种简单的方法,对来自高通量测序管道的原始序列数据进行一些质量控制检查。 它提供了一组模块化的分析,您可以使用这些分析快速了解您的数据是否存在任何问题,在进行任何进一步的分析之前,您应该了解这些问题。 |
|---|---|
| The main functions of FastQC are - Import of data from BAM, SAM or FastQ files (any variant) - Providing a quick overview to tell you in which areas there may be problems - Summary graphs and tables to quickly assess your data - Export of results to an HTML based permanent report - Offline operation to allow automated generation of reports without running the interactive application |
FastQC的主要功能有 - 从BAM、SAM或FastQ文件导入数据(任何变体) - 提供快速概述,告诉您哪些方面可能存在问题 - 用于快速评估数据的摘要图表 - 将结果导出到基于HTML的永久报告 - 脱机操作,无需运行交互式应用程序即可自动生成报告 |
1 Basic Statistics
Summary
The Basic Statistics module generates some simple composition statistics for the file analysed.
- Filename: The original filename of the file which was analysed
- File type: Says whether the file appeared to contain actual base calls or colorspace data which had to be converted to base calls
- Encoding: Says which ASCII encoding of quality values was found in this file.
- Total Sequences: A count of the total number of sequences processed. There are two values reported, actual and estimated. At the moment these will always be the same. In the future it may be possible to analyse just a subset of sequences and estimate the total number, to speed up the analysis, but since we have found that problematic sequences are not evenly distributed through a file we have disabled this for now.
- Filtered Sequences: If running in Casava mode sequences flagged to be filtered will be removed from all analyses. The number of such sequences removed will be reported here. The total sequences count above will not include these filtered sequences and will the number of sequences actually used for the rest of the analysis.
- Sequence Length: Provides the length of the shortest and longest sequence in the set. If all sequences are the same length only one value is reported.
- %GC: The overall %GC of all bases in all sequences
Warning
Basic Statistics never raises a warning.
Failure
Basic Statistics never raises an error.
Common reasons for warnings
This module never raises warnings or errors | 摘要
基本统计模块为所分析的文件生成一些简单的成分统计。
- 文件名:被分析文件的原始文件名
- 文件类型。说明该文件是否包含实际的基础调用或必须转换为基础调用的色彩空间数据
- 编码。说明在此文件中发现了质量值的ASCII编码。
- 总序列。对所处理的序列总数的统计。有两个报告值,实际值和估计值。目前,这些值总是相同的。将来有可能只分析序列的一个子集,并估计总数,以加快分析速度,但由于我们发现有问题的序列在文件中的分布并不均匀,所以我们现在已经禁用了这个功能。
- 过滤的序列:如果在Casava模式下运行,标记为过滤的序列将从所有分析中移除。这里将报告删除的此类序列的数量。上面的总序列数将不包括这些被过滤的序列,而是用于其余分析的实际序列数。
- 序列长度:提供该组中最短和最长的序列的长度。如果所有的序列都是相同的长度,则只报告一个值。
- %GC:所有序列中所有碱基的总体%GC
警告
失败
警告的常见原因
该模块从不产生警告或错误 | | —- | —- |
2 Per Base Sequence Quality
Summary
This view shows an overview of the range of quality values across all bases at each position in the FastQ file. |
摘要
该视图显示了FastQ文件中每个位置的所有碱基的质量值范围的概述。 |
| —- | —- |
| 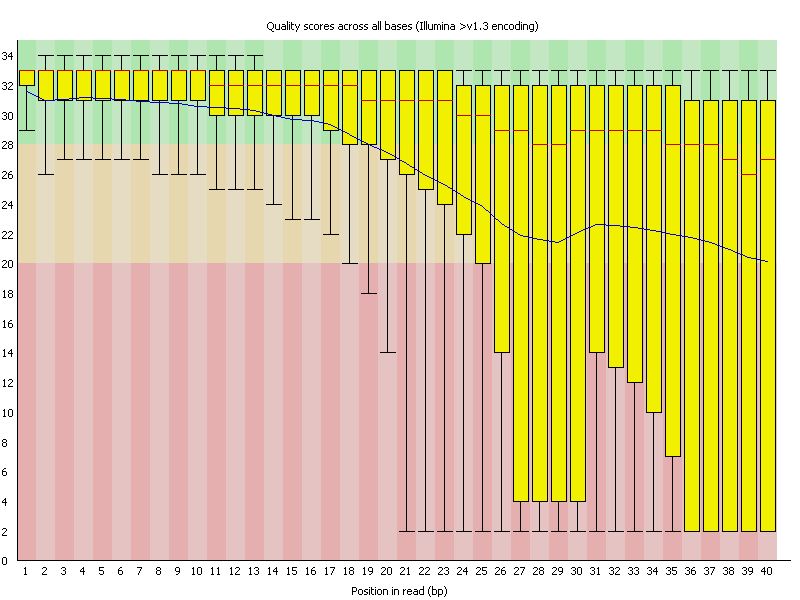 | |
| For each position a BoxWhisker type plot is drawn. The elements of the plot are as follows:
| |
| For each position a BoxWhisker type plot is drawn. The elements of the plot are as follows:
- The central red line is the median value
- The yellow box represents the inter-quartile range (25-75%)
- The upper and lower whiskers represent the 10% and 90% points
- The blue line represents the mean quality
The y-axis on the graph shows the quality scores. The higher the score the better the base call. The background of the graph divides the y axis into very good quality calls (green), calls of reasonable quality (orange), and calls of poor quality (red). The quality of calls on most platforms will degrade as the run progresses, so it is common to see base calls falling into the orange area towards the end of a read.
It should be mentioned that there are number of different ways to encode a quality score in a FastQ file. FastQC attempts to automatically determine which encoding method was used, but in some very limited datasets it is possible that it will guess this incorrectly (ironically only when your data is universally very good!). The title of the graph will describe the encoding FastQC thinks your file used.
Results from this module will not be displayed if your input is a BAM/SAM file in which quality scores have not been recorded. | 对于每个位置,都会画出一个BoxWhisker类型的图。该图的元素如下。
- 中间的红线是中位数值
- 黄色方框代表四分之一区间(25-75%)
- 上方和下方的晶须代表10%和90%的点
- 蓝线代表平均质量
图中的Y轴显示质量分数。分数越高,base call越好。图中的背景将Y轴分为质量非常好的base call(绿色)、质量合理的base call(橙色)和质量差的base call(红色)。
大多数平台上的base call质量会随着运行的进展而下降,所以在read结束时,经常会看到base call落入橙色区域。
应该提到的是,在FastQ文件中,有许多不同的方法来编码质量分数。
FastQC试图自动确定使用的是哪种编码方法,但是在一些非常有限的数据集中,它有可能会猜错(具有讽刺意味的是,只有当您的数据普遍非常好时才会猜错!)。
图表的标题将描述FastQC认为你的文件使用的编码。
如果你的输入是一个没有记录质量分数的BAM/SAM文件,这个模块的结果将不会显示。 |
|
Warning
A warning will be issued if the lower quartile for any base is less than 10, or if the median for any base is less than 25. |
警告
如果任何基数的下四分位数低于10,或任何基数的中位数低于25,就会发出警告。 | |
Failure
This module will raise a failure if the lower quartile for any base is less than 5 or if the median for any base is less than 20. |
失败
如果任何基数的下四分位数小于5或任何基数的中位数小于20,该模块将引发失败。 | |
Common reasons for warnings
The most common reason for warnings and failures in this module is a general degradation of quality over the duration of long runs. In general sequencing chemistry degrades with increasing read length and for long runs you may find that the general quality of the run falls to a level where a warning or error is triggered.
If the quality of the library falls to a low level then the most common remedy is to perform quality trimming where reads are truncated based on their average quality. For most libraries where this type of degradation has occurred you will often be simultaneously running into the issue of adapter read-through so a combined adapter and quality trimming step is often employed.
Another possibility is that a warn / error is triggered because of a short loss of quality earlier in the run, which then recovers to produce later good quality sequence. This can happen if there is a transient problem with the run (bubbles passing through a flowcell for example). You can normally see this type of error by looking at the per-tile quality plot (if available for your platform). In these cases trimming is not advisable as it will remove later good sequence, but you might want to consider masking bases during subsequent mapping or assembly.
If your library has reads of varying length then you can find a warning or error is triggered from this module because of very low coverage for a given base range. Before committing to any action, check how many sequences were responsible for triggering an error by looking at the sequence length distribution module results. |
警告的常见原因
本模块中最常见的警告和失败的原因是在长时间的运行过程中质量普遍下降。
一般来说,测序的化学成分会随着读长的增加而降低,对于长时间的运行,你可能会发现运行的总体质量下降到一个触发警告或错误的水平。
如果文库的质量下降到一个较低的水平,那么最常见的补救措施是进行质量修剪,根据其平均质量将读数截断。
对于大多数发生了这种类型退化的文库,你往往会同时遇到接头读穿的问题,所以通常会采用去接头和质量修剪相结合的步骤。
另一种可能是,由于在运行的早期出现了短暂的质量损失而触发了警告/错误,然后恢复到后来产生的高质量序列。如果运行中出现瞬时问题(例如气泡通过流动池),就会发生这种情况。
通常你可以通过查看每格质量图(如果你的平台上有的话,就是motifs图?)看到这种类型的错误。
在这些情况下,修剪是不可取的,因为它将删除后来的良好序列,但你可能要考虑在随后的mapping或组装过程中对碱基进行屏蔽。
如果你的文库有不同长度的读数,那么你会发现这个模块触发了一个警告或错误,因为对于一个给定的碱基范围,覆盖率非常低。
在承诺采取任何行动之前,通过查看序列长度分布模块的结果,检查有多少序列是负责触发错误的。 |
3 Per Sequence Quality Scores
Summary
The per sequence quality score report allows you to see if a subset of your sequences have universally low quality values. It is often the case that a subset of sequences will have universally poor quality, often because they are poorly imaged (on the edge of the field of view etc), however these should represent only a small percentage of the total sequences. |
摘要
每个序列的质量分数报告允许你查看你的序列的子集是否有普遍的低质量值。
通常情况下,一个序列子集的质量普遍较差,通常是因为它们的成像效果不好(在视野的边缘等),但是这些序列应该只占总序列的一小部分。 |
| —- | —- |
| 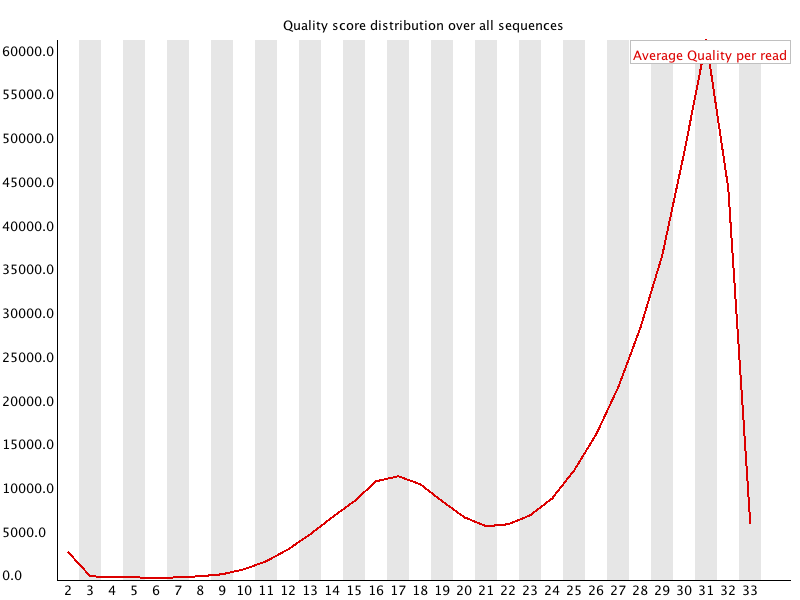 | |
| If a significant proportion of the sequences in a run have overall low quality then this could indicate some kind of systematic problem - possibly with just part of the run (for example one end of a flowcell).
| |
| If a significant proportion of the sequences in a run have overall low quality then this could indicate some kind of systematic problem - possibly with just part of the run (for example one end of a flowcell).
Results from this module will not be displayed if your input is a BAM/SAM file in which quality scores have not been recorded. | 如果运行中相当一部分序列的总体质量较低,那么这可能表明存在某种系统性问题—可能只是运行的一部分(例如流动池的一端)。
如果你的输入是一个没有记录质量分数的BAM/SAM文件,这个模块的结果将不会显示。 |
|
Warning
A warning is raised if the most frequently observed mean quality is below 27 - this equates to a 0.2% error rate. |
警告
如果最经常观察到的平均质量低于27分,就会发出警告—这相当于0.2%的错误率。 | |
Failure
An error is raised if the most frequently observed mean quality is below 20 - this equates to a 1% error rate. |
失败
如果最经常观察到的平均质量低于20,就会出现错误,这相当于1%的错误率。 | |
Common reasons for warnings
This module is generally fairly robust and errors here usually indicate a general loss of quality within a run. For long runs this may be alleviated through quality trimming. If a bi-modal, or complex distribution is seen then the results should be evaluated in concert with the per-tile qualities (if available) since this might indicate the reason for the loss in quality of a subset of sequences. |
警告的常见原因
这个模块通常是相当稳健的,这里的错误通常表明在一个运行中质量的普遍损失。
对于长时间的运行,这可以通过质量修剪来缓解。如果出现双模或复杂的分布,那么应该与每格质量(如果有的话)一起评估结果,因为这可能表明序列子集质量损失的原因。 |
4 Per Base Sequence Content
Summary
Per Base Sequence Content plots out the proportion of each base position in a file for which each of the four normal DNA bases has been called. |
摘要
每个碱基序列含量绘制出文件中每个碱基位置的比例,其中四个正常的DNA碱基都被call。 |
| —- | —- |
| 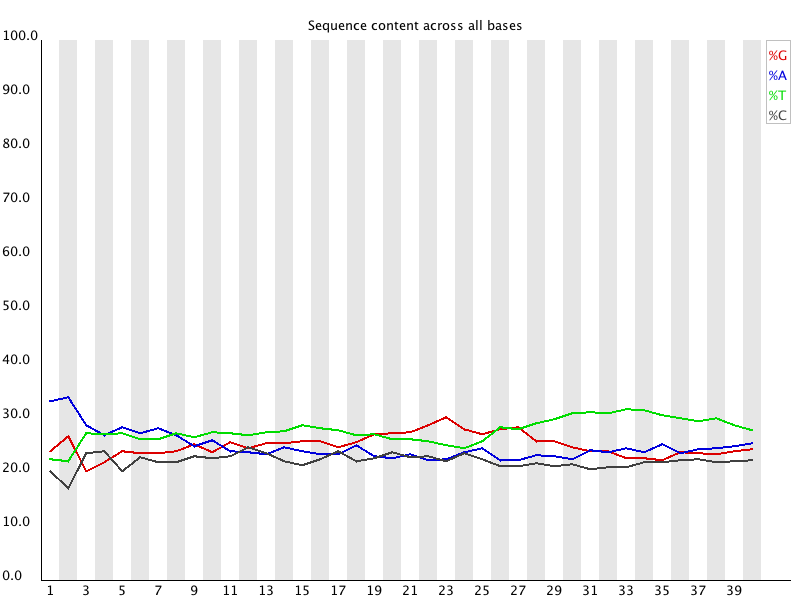 | |
| In a random library you would expect that there would be little to no difference between the different bases of a sequence run, so the lines in this plot should run parallel with each other. The relative amount of each base should reflect the overall amount of these bases in your genome, but in any case they should not be hugely imbalanced from each other.
| |
| In a random library you would expect that there would be little to no difference between the different bases of a sequence run, so the lines in this plot should run parallel with each other. The relative amount of each base should reflect the overall amount of these bases in your genome, but in any case they should not be hugely imbalanced from each other.
It’s worth noting that some types of library will always produce biased sequence composition, normally at the start of the read. Libraries produced by priming using random hexamers (including nearly all RNA-Seq libraries) and those which were fragmented using transposases inherit an intrinsic bias in the positions at which reads start. This bias does not concern an absolute sequence, but instead provides enrichement of a number of different K-mers at the 5’ end of the reads. Whilst this is a true technical bias, it isn’t something which can be corrected by trimming and in most cases doesn’t seem to adversely affect the downstream analysis. It will however produce a warning or error in this module. | 在一个随机文库中,你会发现一个序列的不同碱基之间几乎没有差别,所以这个图中的线条应该是相互平行的。
每个碱基的相对数量应该反映出这些碱基在你的基因组中的总体数量,但在任何情况下,它们之间不应该有巨大的不平衡。
值得注意的是,某些类型的文库总是会产生有偏差的序列组成,通常是在read的开始。
使用随机六聚体引物产生的文库(包括几乎所有的RNA-Seq文库)和使用转座酶分割的文库在read开始的位置上都有内在的偏差。
这种偏差并不涉及绝对序列,而是在读数的5’端提供一些不同的K-mers的富集。
虽然这是一个真正的技术偏差,但它不是可以通过修剪来纠正的,而且在大多数情况下似乎不会对下游的分析产生不利影响。但是在这个模块中会产生一个警告或错误。 |
|
Warning
This module issues a warning if the difference between A and T, or G and C is greater than 10% in any position. |
警告
如果A和T或G和C之间的差异在任何位置上都大于10%,本模块就会发出警告。 | |
Failure
This module will fail if the difference between A and T, or G and C is greater than 20% in any position. |
失败
如果A和T,或G和C之间的差异在任何位置上大于20%,本模块将失败。 | |
Common reasons for warnings
There are a number of common scenarios which would ellicit a warning or error from this module.
1. Overrepresented sequences: If there is any evidence of overrepresented sequences such as adapter dimers or rRNA in a sample then these sequences may bias the overall composition and their sequence will emerge from this plot.
1. Biased fragmentation: Any library which is generated based on the ligation of random hexamers or through tagmentation should theoretically have good diversity through the sequence, but experience has shown that these libraries always have a selection bias in around the first 12bp of each run. This is due to a biased selection of random primers, but doesn’t represent any individually biased sequences. Nearly all RNA-Seq libraries will fail this module because of this bias, but this is not a problem which can be fixed by processing, and it doesn’t seem to adversely affect the ablity to measure expression.
1. Biased composition libraries: Some libraries are inherently biased in their sequence composition. The most obvious example would be a library which has been treated with sodium bisulphite which will then have converted most of the cytosines to thymines, meaning that the base composition will be almost devoid of cytosines and will thus trigger an error, despite this being entirely normal for that type of library
1. If you are analysing a library which has been aggressivley adapter trimmed then you will naturally introduce a composition bias at the end of the reads as sequences which happen to match short stretches of adapter are removed, leaving only sequences which do not match. Sudden deviations in composition at the end of libraries which have undergone aggressive trimming are therefore likely to be spurious.
|
发出警告的常见原因
有一些常见的情况会引起本模块的警告或错误。
1. 代表性过强的序列:如果有任何证据表明样品中存在过度代表的序列,如接头二聚体或rRNA,那么这些序列可能偏向于整体组成,其序列将从该图中出现。
2. 有偏见的片段:任何基于随机六聚体连接或通过标记产生的文库,理论上都应该有良好的序列多样性,但经验表明,这些文库在每次运行的前12bp左右总是有选择偏差。
这是由于随机引物的选择有偏差,但并不代表任何单独有偏差的序列。几乎所有的RNA-Seq文库都会因为这种偏差而不能通过这个模块,但这不是一个可以通过处理来解决的问题,而且它似乎不会对测量表达的能力产生不利的影响。
3. 有偏见的组成库:有些文库在其序列组成上有内在的偏差。最明显的例子是用亚硫酸钠处理过的文库,它将大部分的胞嘧啶转化为胸腺嘧啶,这意味着碱基组成几乎没有胞嘧啶,因此会引发错误,尽管这对该类型的文库来说是完全正常的。
4. 如果你正在分析一个被过度修剪过的接头文库,那么你自然会在read的末端引入一个成分偏差,因为碰巧与短的接头相匹配的序列被移除,只留下不匹配的序列。因此,在经过积极修剪的文库末端突然出现的成分偏差很可能是虚假的。
|
5 Per Sequence GC Content
Summary
This module measures the GC content across the whole length of each sequence in a file and compares it to a modelled normal distribution of GC content. |
摘要
该模块测量文件中每个序列的整个长度的GC含量,并将其与GC含量的模拟正态分布进行比较。 |
| —- | —- |
| 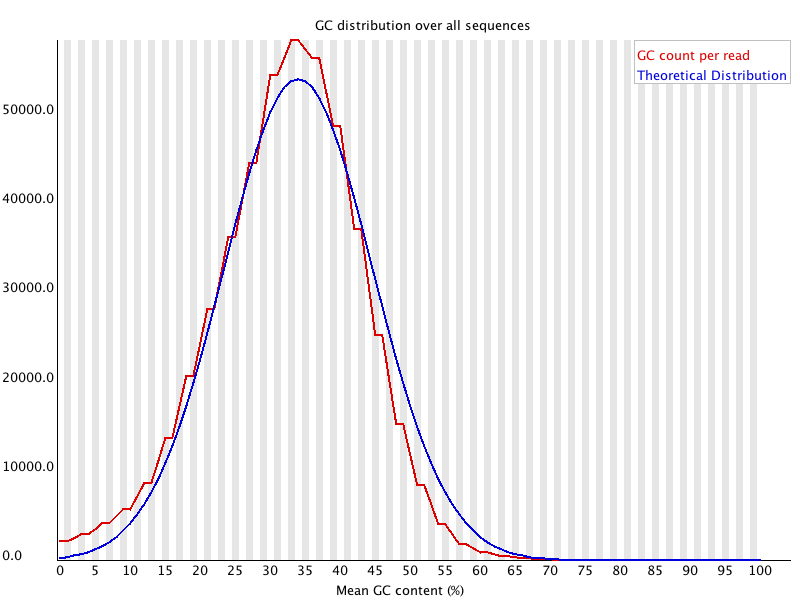 | |
| In a normal random library you would expect to see a roughly normal distribution of GC content where the central peak corresponds to the overall GC content of the underlying genome. Since we don’t know the the GC content of the genome the modal GC content is calculated from the observed data and used to build a reference distribution.
| |
| In a normal random library you would expect to see a roughly normal distribution of GC content where the central peak corresponds to the overall GC content of the underlying genome. Since we don’t know the the GC content of the genome the modal GC content is calculated from the observed data and used to build a reference distribution.
An unusually shaped distribution could indicate a contaminated library or some other kinds of biased subset. A normal distribution which is shifted indicates some systematic bias which is independent of base position. If there is a systematic bias which creates a shifted normal distribution then this won’t be flagged as an error by the module since it doesn’t know what your genome’s GC content should be. | 在一个正常的随机库中,你会期望看到一个大致正常的GC含量分布,其中中心峰对应于基础基因组的整体GC含量。由于我们不知道基因组的GC含量,所以根据观察到的数据计算出GC含量的模数,并用来建立一个参考分布。
一个不寻常的分布可能表示一个被污染的文库或其他类型的有偏见的子集。
一个偏移的正态分布表明有一些与碱基位置无关的系统偏差。如果有一个系统性的偏差造成了一个偏移的正态分布,那么这不会被模块标记为一个错误,因为它不知道你的基因组的GC含量应该是什么。 |
|
Warning
A warning is raised if the sum of the deviations from the normal distribution represents more than 15% of the reads. |
警告
如果偏离正态分布的总和占读数的15%以上,则发出警告。 | |
Failure
This module will indicate a failure if the sum of the deviations from the normal distribution represents more than 30% of the reads. |
失败
如果偏离正常分布的总和占读数的30%以上,该模块将提示失败。 | |
Common reasons for warnings
Warnings in this module usually indicate a problem with the library. Sharp peaks on an otherwise smooth distribution are normally the result of a specific contaminant (adapter dimers for example), which may well be picked up by the overrepresented sequences module. Broader peaks may represent contamination with a different species. |
警告的常见原因
本模块中的警告通常表明文库有问题。平滑分布上的尖锐峰值通常是特定污染物的结果(例如接头二聚体),这很可能被过度代表的序列模块所发现。
较宽的峰值可能代表不同物种的污染。 |
6 Per Base N Content
Summary
If a sequencer is unable to make a base call with sufficient confidence then it will normally substitute an N rather than a conventional base call
This module plots out the percentage of base calls at each position for which an N was called. | 总结
如果测序仪无法以足够的置信度进行base call,则它通常会替换N而不是传统的base call
此模块绘制了在每个被识别为N的位置的base call百分比 |
| —- | —- |
| 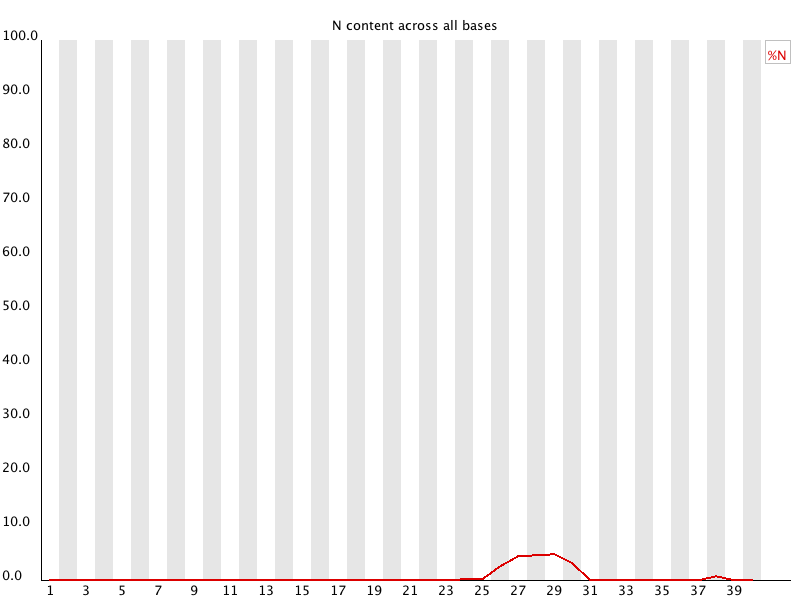 | |
| It’s not unusual to see a very low proportion of Ns appearing in a sequence, especially nearer the end of a sequence. However, if this proportion rises above a few percent it suggests that the analysis pipeline was unable to interpret the data well enough to make valid base calls. | 在一个序列中出现非常低的N的比例是很正常的,特别是在接近序列的末端。
| |
| It’s not unusual to see a very low proportion of Ns appearing in a sequence, especially nearer the end of a sequence. However, if this proportion rises above a few percent it suggests that the analysis pipeline was unable to interpret the data well enough to make valid base calls. | 在一个序列中出现非常低的N的比例是很正常的,特别是在接近序列的末端。
然而,如果这个比例上升到百分之几以上,就表明分析流程不能很好地解释数据,不能做出有效的base call。 |
|
Warning
This module raises a warning if any position shows an N content of >5%. |
警告
Failure
This module will raise an error if any position shows an N content of >20%. |
失败
如果任何位置显示N含量>20%,该模块将引发一个错误。 | |
Common reasons for warnings
The most common reason for the inclusion of significant proportions of Ns is a general loss of quality, so the results of this module should be evaluated in concert with those of the various quality modules. You should check the coverage of a specific bin, since it’s possible that the last bin in this analysis could contain very few sequences, and an error could be prematurely triggered in this case.
Another common scenario is the incidence of a high proportions of N at a small number of positions early in the library, against a background of generally good quality. Such deviations can occur when you have very biased sequence composition in the library to the point that base callers can become confused and make poor calls. This type of problem will be apparent when looking at the per-base sequence content results. |
警告的常见原因
含有大量N的最常见的原因是质量的普遍损失,所以这个模块的结果应该和各种质量模块的结果一起评估。
你应该检查特定仓的覆盖率,因为这个分析中的最后一个仓有可能包含很少的序列,在这种情况下可能会过早地触发错误。
另一种常见的情况是,在库中早期的少数位置上出现了高比例的N,而背景是质量普遍良好。
当你在文库中的序列组成有很大的偏差,以至于碱基调用者会感到困惑并做出错误的call时,就会出现这种偏差。
这种类型的问题在查看每个碱基的序列含量结果时将会很明显。 |
7 Sequence Length Distribution
Summary
Some high throughput sequencers generate sequence fragments of uniform length, but others can contain reads of wildly varying lengths. Even within uniform length libraries some pipelines will trim sequences to remove poor quality base calls from the end.
This module generates a graph showing the distribution of fragment sizes in the file which was analysed. |
摘要
一些高通量测序仪产生统一长度的序列片段,但其他测序仪可能包含长度大不相同的读数。
即使在统一长度的库中,一些流程也会修剪序列以去除末端的劣质base call。
该模块生成一个图表,显示被分析的文件中片段大小的分布。 |
| —- | —- |
| 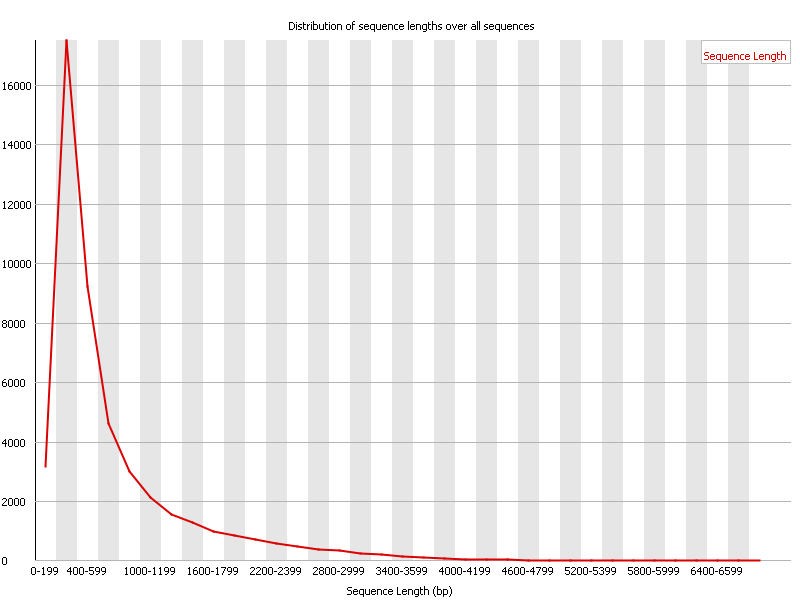 | |
| In many cases this will produce a simple graph showing a peak only at one size, but for variable length FastQ files this will show the relative amounts of each different size of sequence fragment. | 在许多情况下,这将产生一个简单的图表,只显示一个大小的峰值,但对于可变长度的FastQ文件,这将显示每个不同大小的序列片段的相对数量。 |
|
| |
| In many cases this will produce a simple graph showing a peak only at one size, but for variable length FastQ files this will show the relative amounts of each different size of sequence fragment. | 在许多情况下,这将产生一个简单的图表,只显示一个大小的峰值,但对于可变长度的FastQ文件,这将显示每个不同大小的序列片段的相对数量。 |
|
Warning
This module will raise a warning if all sequences are not the same length. |
警告
Failure
This module will raise an error if any of the sequences have zero length. |
失败
Common reasons for warnings
For some sequencing platforms it is entirely normal to have different read lengths so warnings here can be ignored. |
警告的常见原因
对于一些测序平台来说,有不同的读长度是完全正常的,所以这里的警告可以被忽略。 |
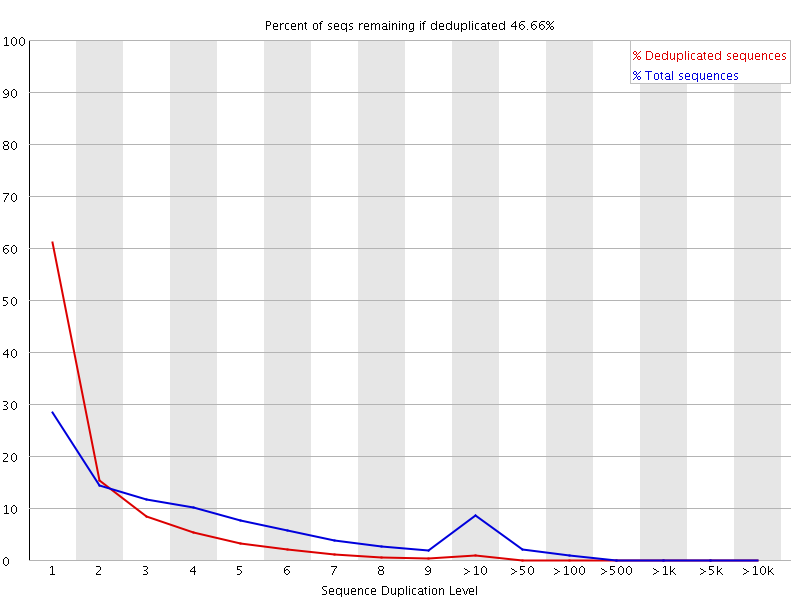
8 Duplicate Sequences
Summary
In a diverse library most sequences will occur only once in the final set. A low level of duplication may indicate a very high level of coverage of the target sequence, but a high level of duplication is more likely to indicate some kind of enrichment bias (eg PCR over amplification).
This module counts the degree of duplication for every sequence in a library and creates a plot showing the relative number of sequences with different degrees of duplication. |
摘要
在一个多样化的文库中,大多数序列在最后的集合中只出现一次。低水平的重复可能表明目标序列的覆盖率非常高,但高水平的重复更可能表明某种富集偏差(如PCR过度扩增)。
该模块计算文库中每个序列的重复程度,并创建一个图表,显示具有不同重复程度的序列的相对数量。 |
| —- | —- |
|  | |
| To cut down on the memory requirements for this module only sequences which first appear in the first 100,000 sequences in each file are analysed, but this should be enough to get a good impression for the duplication levels in the whole file. Each sequence is tracked to the end of the file to give a representative count of the overall duplication level. To cut down on the amount of information in the final plot any sequences with more than 10 duplicates are placed into grouped bins to give a clear impression of the overall duplication level without having to show each individual duplication value. | 为了减少该模块的内存需求,只分析每个文件中首次出现的100,000个序列,但这应该足以对整个文件的重复水平有一个好的印象。
| |
| To cut down on the memory requirements for this module only sequences which first appear in the first 100,000 sequences in each file are analysed, but this should be enough to get a good impression for the duplication levels in the whole file. Each sequence is tracked to the end of the file to give a representative count of the overall duplication level. To cut down on the amount of information in the final plot any sequences with more than 10 duplicates are placed into grouped bins to give a clear impression of the overall duplication level without having to show each individual duplication value. | 为了减少该模块的内存需求,只分析每个文件中首次出现的100,000个序列,但这应该足以对整个文件的重复水平有一个好的印象。
每个序列都被追踪到文件的末尾,以提供总体重复水平的代表性计数。
为了减少最终图表中的信息量,任何有10个以上重复的序列都被放在分组的仓中,以便对整体的重复水平有一个清晰的印象,而不需要显示每个单独的重复值。 |
| Because the duplication detection requires an exact sequence match over the whole length of the sequence, any reads over 75bp in length are truncated to 50bp for the purposes of this analysis. Even so, longer reads are more likely to contain sequencing errors which will artificially increase the observed diversity and will tend to underrepresent highly duplicated sequences. | 由于重复检测需要在整个序列的长度上进行精确的序列匹配,因此在本分析中,任何长度超过75bp的读数被截断为50bp。
即便如此,较长的读数更有可能包含测序错误,这将人为地增加观察到的多样性,并倾向于对高度重复的序列的代表不足。 |
| The plot shows the proportion of the library which is made up of sequences in each of the different duplication level bins. There are two lines on the plot. The blue line takes the full sequence set and shows how its duplication levels are distributed. In the red plot the sequences are de-duplicated and the proportions shown are the proportions of the deduplicated set which come from different duplication levels in the original data. | 该图显示了在每个不同的复制水平区间内,由序列组成的库的比例。
该图上有两条线。蓝线是完整的序列集,显示其复制水平的分布。在红色图中,序列被去掉了重复,显示的比例是来自原始数据中不同重复水平的重复集的比例。 |
| In a properly diverse library most sequences should fall into the far left of the plot in both the red and blue lines. A general level of enrichment, indicating broad oversequencing in the library will tend to flatten the lines, lowering the low end and generally raising other categories. More specific enrichments of subsets, or the presence of low complexity contaminants will tend to produce spikes towards the right of the plot. These high duplication peaks will most often appear in the blue trace as they make up a high proportion of the original library, but usually disappear in the red trace as they make up an insignificant proportion of the deduplicated set. If peaks persist in the blue trace then this suggests that there are a large number of different highly duplicated sequences which might indicate either a contaminant set or a very severe technical duplication. | 在一个适当的多样化文库中,大多数序列应该落在图中红线和蓝线的最左边。
一般水平的富集,表明库中广泛的过度测序,将倾向于使线条变平,降低低端,一般提高其他类别。
更具体的子集富集,或存在低复杂度的污染物,将倾向于在图的右边产生尖峰。
这些高重复度的峰值往往会出现在蓝色的图谱中,因为它们在原始库中占了很高的比例,但通常会在红色的图谱中消失,因为它们在重复数据集中占的比例不大。
如果峰持续出现在蓝色轨迹中,那么这表明有大量不同的高度重复的序列,这可能表明有污染的序列或非常严重的技术重复。 |
| The module also calculates an expected overall loss of sequence were the library to be deduplicated. This headline figure is shown at the top of the plot and gives a reasonable impression of the potential overall level of loss. | 该模块还计算了文库被重复复制后的预期整体序列损失。
这个标题数字显示在图表的顶部,对潜在的整体损失水平给出了合理的印象。 |
|
Warning
This module will issue a warning if non-unique sequences make up more than 20% of the total. |
警告
如果非唯一序列占总数的20%以上,该模块将发出警告。 | |
Failure
This module will issue a error if non-unique sequences make up more than 50% of the total. |
失败
如果非唯一序列占总数的50%以上,该模块将发出一个错误。 | |
Common reasons for warnings
The underlying assumption of this module is of a diverse unenriched library. Any deviation from this assumption will naturally generate duplicates and can lead to warnings or errors from this module.
In general there are two potential types of duplicate in a library, technical duplicates arising from PCR artefacts, or biological duplicates which are natural collisions where different copies of exactly the same sequence are randomly selected. From a sequence level there is no way to distinguish between these two types and both will be reported as duplicates here.
A warning or error in this module is simply a statement that you have exhausted the diversity in at least part of your library and are re-sequencing the same sequences. In a supposedly diverse library this would suggest that the diversity has been partially or completely exhausted and that you are therefore wasting sequencing capacity. However in some library types you will naturally tend to over-sequence parts of the library and therefore generate duplication and will therefore expect to see warnings or error from this module.
In RNA-Seq libraries sequences from different transcripts will be present at wildly different levels in the starting population. In order to be able to observe lowly expressed transcripts it is therefore common to greatly over-sequence high expressed transcripts, and this will potentially create large set of duplicates. This will result in high overall duplication in this test, and will often produce peaks in the higher duplication bins. This duplication will come from physically connected regions, and an examination of the distribution of duplicates in a specific genomic region will allow the distinction between over-sequencing and general technical duplication, but these distinctions are not possible from raw fastq files. A similar situation can arise in highly enriched ChIP-Seq libraries although the duplication there is less pronounced. Finally, if you have a library where the sequence start points are constrained (a library constructed around restriction sites for example, or an unfragmented small RNA library) then the constrained start sites will generate huge dupliction levels which should not be treated as a problem, nor removed by deduplication. In these types of library you should consider using a system such as random barcoding to allow the distinction of technical and biological duplicates. |
出现警告的常见原因
本模块的基本假设是一个多样化的非富集库。任何偏离这一假设的行为都会自然产生重复,并可能导致本模块出现警告或错误。
一般来说,库中有两种潜在的重复类型,一种是由PCR伪装引起的技术性重复,另一种是生物性重复,即完全相同的序列的不同拷贝被随机选择的自然碰撞。从序列层面上看,没有办法区分这两种类型,因此这两种类型在这里都将被报告为重复。
这个模块中的警告或错误只是说明你已经用尽了库中至少一部分的多样性,正在对相同的序列进行重新测序。
在一个所谓的多样性文库中,这将表明多样性已经部分或完全用尽,因此你正在浪费测序能力。
然而,在一些文库类型中,你会自然而然地倾向于对部分文库进行过度测序,从而产生重复,因此会期望从这个模块看到警告或错误。
在RNA-Seq文库中,来自不同转录本的序列在起始群体中的存在程度大不相同。
为了能够观察到低表达的转录本,通常会对高表达的转录本进行大量的过度测序,这将有可能产生大量的重复。
这将导致本试验中的总体重复率很高,并经常在较高的重复率区产生峰值。
这种重复将来自于物理上相连的区域,对特定基因组区域的重复分布的检查将允许区分过度测序和一般的技术性重复,但这些区分不可能从原始fastq文件中得到。
类似的情况也会出现在高度富集的ChIP-Seq文库中,尽管那里的重复没有那么明显。
最后,如果你有一个序列起始点受到限制的文库(例如围绕限制性位点构建的文库,或一个未破碎的小RNA文库),那么受限制的起始点将产生巨大的重复水平,这不应该被视为一个问题,也不能通过重复数据删除来消除。
在这些类型的文库中,你应该考虑使用一个系统,如随机条码,以区分技术和生物重复。 |
9 Overrepresented Sequences
Summary
A normal high-throughput library will contain a diverse set of sequences, with no individual sequence making up a tiny fraction of the whole. Finding that a single sequence is very overrepresented in the set either means that it is highly biologically significant, or indicates that the library is contaminated, or not as diverse as you expected. |
摘要
一个正常的高通量文库将包含一个多样化的序列集,没有一个单独的序列只占整个序列的一小部分。
如果发现一个单一的序列在整个库中占的比例过高,这意味着它具有高度的生物学意义,或者表明库被污染了,或者不像你预期的那样多样化。 |
| —- | —- |
| This module lists all of the sequence which make up more than 0.1% of the total. To conserve memory only sequences which appear in the first 100,000 sequences are tracked to the end of the file. It is therefore possible that a sequence which is overrepresented but doesn’t appear at the start of the file for some reason could be missed by this module. | 这个模块列出了所有占总数0.1%以上的序列。为了节省内存,只有出现在前10万个序列中的序列被追踪到文件的末尾。
因此,由于某种原因没有出现在文件开始的序列有可能被这个模块所遗漏,它是超比例的。 |
| For each overrepresented sequence the program will look for matches in a database of common contaminants and will report the best hit it finds. Hits must be at least 20bp in length and have no more than 1 mismatch. Finding a hit doesn’t necessarily mean that this is the source of the contamination, but may point you in the right direction. It’s also worth pointing out that many adapter sequences are very similar to each other so you may get a hit reported which isn’t technically correct, but which has very similar sequence to the actual match. | 对于每一个高代表度的序列,该程序将在常见污染物的数据库中寻找匹配的序列,并报告它所发现的最佳命中。
命中必须至少有20bp的长度,并且不超过1个错配。找到一个命中不一定意味着这就是污染源,但可以为你指出正确的方向。还值得指出的是,许多适配器序列彼此非常相似,所以你可能得到一个技术上不正确的命中,但它的序列与实际的匹配非常相似。 |
| Because the duplication detection requires an exact sequence match over the whole length of the sequence any reads over 75bp in length are truncated to 50bp for the purposes of this analysis. Even so, longer reads are more likely to contain sequencing errors which will artificially increase the observed diversity and will tend to underrepresent highly duplicated sequences. | 因为重复检测需要在整个序列的长度上有一个精确的序列匹配,所以为了这个分析的目的,超过75bp的读数被截断为50bp。即便如此,较长的读数更有可能包含测序错误,这将人为地增加观察到的多样性,并倾向于低估高度重复的序列的代表性。 |
|
Warning
This module will issue a warning if any sequence is found to represent more than 0.1% of the total. |
警告
如果发现任何序列占总数的0.1%以上,该模块将发出警告。 | |
Failure
This module will issue an error if any sequence is found to represent more than 1% of the total. |
失败
如果发现任何序列占总数的1%以上,该模块将发出一个错误。 | |
Common reasons for warnings
This module will often be triggered when used to analyse small RNA libraries where sequences are not subjected to random fragmentation, and the same sequence may natrually be present in a significant proportion of the library. |
警告的常见原因
当用于分析小的RNA文库时,该模块经常被触发,在这些文库中,序列不受随机破碎的影响,相同的序列可能自然地存在于文库的很大一部分。 |
10 Adapter Content
Summary
The Kmer Content module will do a generic analysis of all of the Kmers in your library to find those which do not have even coverage through the length of your reads. This can find a number of different sources of bias in the library which can include the presence of read-through adapter sequences building up on the end of your sequences.
You can however find that the presence of any overrepresented sequences in your library (such as adapter dimers) will cause the Kmer plot to be dominated by the Kmers these sequences contain, and that it’s not always easy to see if there are other biases present in which you might be interested.
One obvious class of sequences which you might want to analyse are adapter sequences. It is useful to know if your library contains a significant amount of adapter in order to be able to assess whether you need to adapter trim or not. Although the Kmer analysis can theoretically spot this kind of contamination it isn’t always clear. This module therefore does a specific search for a set of separately defined Kmers and will give you a view of the total proportion of your library which contain these Kmers. A results trace will always be generated for all of the sequences present in the adapter config file so you can see the adapter content of your library, even if it’s low.
The plot itself shows a cumulative percentage count of the proportion of your library which has seen each of the adapter sequences at each position. Once a sequence has been seen in a read it is counted as being present right through to the end of the read so the percentages you see will only increase as the read length goes on. |
摘要
Kmer Content模块将对你库中的所有Kmer进行通用分析,找出那些在读数长度上没有均匀覆盖的Kmer。这可以在文库中找到一些不同的偏差来源,其中包括在你的序列末端出现的通读接头序列。
然而,你可以发现在你的文库中存在任何过度代表的序列(如接头二聚体)将导致Kmer图被这些序列所包含的Kmer所支配,而且并不总是容易看到是否存在你可能感兴趣的其他偏倚。
你可能想分析的一类明显的序列是接头序列。知道你的文库是否含有大量的接头是很有用的,以便能够评估你是否需要进行接头修剪。
尽管Kmer分析理论上可以发现这种污染,但并不总是很清楚。因此,这个模块对一组单独定义的Kmers进行特定的搜索,并将给你一个包含这些Kmers的库的总比例的视图。
一个结果跟踪将总是为所有存在于接头配置文件中的序列生成,所以你可以看到你的库中的适配器含量,即使它很低。
该图本身显示了你的文库在每个位置上看到每个接头序列的比例的累积百分比计数。一旦一个序列在read中出现,它就会被计算为一直存在到read的末端,所以你看到的百分比只会随着读数长度的增加而增加。 |
| —- | —- |
|
Warning
This module will issue a warning if any sequence is present in more than 5% of all reads.
Failure
This module will issue a warning if any sequence is present in more than 10% of all reads.
Common reasons for warnings
Any library where a reasonable proportion of the insert sizes are shorter than the read length will trigger this module. This doesn’t indicate a problem as such - just that the sequences will need to be adapter trimmed before proceeding with any downstream analysis. |
警告
如果任何序列出现在所有read中的5%以上,该模块将发出警告。
失败
如果任何序列出现在所有read的10%以上,该模块将发出警告。
警告的常见原因
任何库中,如果有合理比例的插入物尺寸短于读数长度,将触发该模块。这并不表明有什么问题—只是在进行任何下游分析之前,需要对序列进行转接器修剪。 |
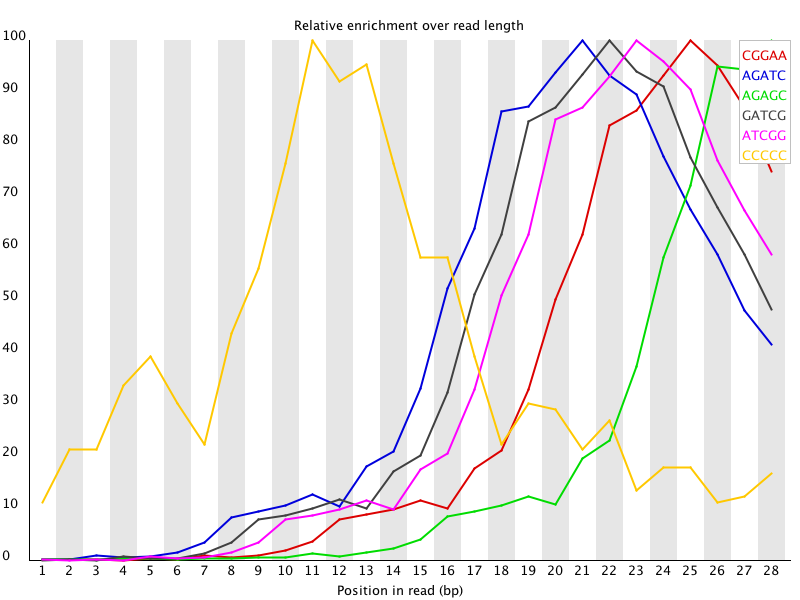
11 Kmer Content
Summary
The analysis of overrepresented sequences will spot an increase in any exactly duplicated sequences, but there are a different subset of problems where it will not work.
- If you have very long sequences with poor sequence quality then random sequencing errors will dramatically reduce the counts for exactly duplicated sequences.
- If you have a partial sequence which is appearing at a variety of places within your sequence then this won’t be seen either by the per base content plot or the duplicate sequence analysis.
| 摘要
过度代表序列的分析将发现任何完全重复的序列的增加,但有一个不同的问题子集,它将不起作用。
- 如果你有很长的序列,序列质量很差,那么随机测序错误将大大减少完全重复序列的数量。
- 如果你有一个部分序列在你的序列中出现在不同的地方,那么这将不会被每个碱基含量图或重复序列分析所看到。
|
| —- | —- |
| The Kmer module starts from the assumption that any small fragment of sequence should not have a positional bias in its apearance within a diverse library. There may be biological reasons why certain Kmers are enriched or depleted overall, but these biases should affect all positions within a sequence equally. This module therefore measures the number of each 7-mer at each position in your library and then uses a binomial test to look for significant deviations from an even coverage at all positions. Any Kmers with positionally biased enrichment are reported. The top 6 most biased Kmer are additionally plotted to show their distribution. | Kmer模块的出发点是,任何小的序列片段在不同的文库中出现时都不应该有位置上的偏差。
可能有生物学上的原因导致某些Kmer在整体上被富集或耗尽,但这些偏倚应该平等地影响序列中的所有位置。
因此,该模块测量库中每个位置上的每个7-mer的数量,然后使用二项式测试来寻找所有位置上的均匀覆盖的显著偏差。
任何有位置偏向性富集的Kmer都会被报告。另外,还绘制了前6个最偏向的Kmer,以显示其分布。 |
|  | |
| To allow this module to run in a reasonable time only 2% of the whole library is analysed and the results are extrapolated to the rest of the library. Sequences longer than 500bp are truncated to 500bp for this analysis. | 为了使这个模块能在合理的时间内运行,只对整个文库的2%进行分析,并将结果推断到文库的其他部分。长于500bp的序列在此分析中被截断为500bp。 |
|
| |
| To allow this module to run in a reasonable time only 2% of the whole library is analysed and the results are extrapolated to the rest of the library. Sequences longer than 500bp are truncated to 500bp for this analysis. | 为了使这个模块能在合理的时间内运行,只对整个文库的2%进行分析,并将结果推断到文库的其他部分。长于500bp的序列在此分析中被截断为500bp。 |
|
Warning
This module will issue a warning if any k-mer is imbalanced with a binomial p-value <0.01.
Failure
This module will issue a warning if any k-mer is imbalanced with a binomial p-value < 10^-5.
Common reasons for warnings
Any individually overrepresented sequences, even if not present at a high enough threshold to trigger the overrepresented sequences module will cause the Kmers from those sequences to be highly enriched in this module. These will normally appear as sharp spikes of enrichemnt at a single point in the sequence, rather than a progressive or broad enrichment.
Libraries which derive from random priming will nearly always show Kmer bias at the start of the library due to an incomplete sampling of the possible random primers. |
警告
如果任何K-mer不平衡,二项式P值小于0.01,该模块将发出警告。
失败
如果任何k-mer不平衡,二项式p值<10^-5,本模块将发出警告。
警告的常见原因
任何单独的过度代表的序列,即使没有达到足够高的阈值来触发过度代表的序列模块,也会导致这些序列的Kmers在这个模块中高度富集。这些通常会在序列的一个点上出现尖锐的富集,而不是渐进的或广泛的富集。
由于对可能的随机引物的不完全抽样,来自随机引物的文库几乎总是在文库的开始出现Kmer的偏差。
|
12 Per Tile Sequence Quality
Summary
This graph will only appear in your analysis results if you’re using an Illumina library which retains its original sequence identifiers. Encoded in these is the flowcell tile from which each read came. The graph allows you to look at the quality scores from each tile across all of your bases to see if there was a loss in quality associated with only one part of the flowcell. |
摘要
只有当你使用的Illumina文库保留了其原始序列标识符时,该图才会出现在你的分析结果中。
编码在这些标识中的是每个读数来自的流室瓦片。该图允许你查看每个瓦片在所有碱基上的质量得分,以了解是否有质量损失只与flowcell的一个部分有关。 |
| —- | —- |
| The plot shows the deviation from the average quality for each tile. The colours are on a cold to hot scale, with cold colours being positions where the quality was at or above the average for that base in the run, and hotter colours indicate that a tile had worse qualities than other tiles for that base. In the example below you can see that certain tiles show consistently poor quality. A good plot should be blue all over. | 该图显示了每一瓦片与平均质量的偏差。颜色是由冷到热的,冷色是指在运行中质量达到或超过该基地的平均水平的位置,较热的颜色表示一个瓷砖的质量比该基地的其他瓷砖差。在下面的例子中,你可以看到某些瓷砖的质量一直很差。一个好的地块应该是全部都是蓝色的。 |
|  | |
| Reasons for seeing warnings or errors on this plot could be transient problems such as bubbles going through the flowcell, or they could be more permanent problems such as smudges on the flowcell or debris inside the flowcell lane. | 在这个图上看到警告或错误的原因可能是短暂的问题,如气泡穿过流动池,也可能是更持久的问题,如流动池上的污点或流动池通道内的碎片。 |
|
| |
| Reasons for seeing warnings or errors on this plot could be transient problems such as bubbles going through the flowcell, or they could be more permanent problems such as smudges on the flowcell or debris inside the flowcell lane. | 在这个图上看到警告或错误的原因可能是短暂的问题,如气泡穿过流动池,也可能是更持久的问题,如流动池上的污点或流动池通道内的碎片。 |
|
Warning
This module will issue a warning if any tile shows a mean Phred score more than 2 less than the mean for that base across all tiles.
Failure
This module will issue a warning if any tile shows a mean Phred score more than 5 less than the mean for that base across all tiles.
Common reasons for warnings
Whilst warnings in this module can be triggered by individual specific events we have also observed that greater variation in the phred scores attributed to tiles can also appear when a flowcell is generally overloaded. In this case events appear all over the flowcell rather than being confined to a specific area or range of cycles. We would generally ignore errors which mildly affected a small number of tiles for only 1 or 2 cycles, but would pursue larger effects which showed high deviation in scores, or which persisted for several cycles. |
警告
如果任何瓦片显示的平均Phred分数比该基地所有瓦片的平均分数低2以上,该模块将发出警告。
失败
如果任何瓦片显示的Phred平均分比该基地所有瓦片的平均分低5分以上,该模块将发出一个警告。
警告的常见原因
虽然本模块的警告可以由个别特定事件触发,但我们也观察到,当一个流动池普遍过载时,归于瓦片的Phred分数也会出现较大变化。在这种情况下,事件出现在整个流程单元,而不是局限于一个特定的区域或周期范围。我们一般会忽略那些只对1或2个周期的少量瓷砖产生轻微影响的错误,但会追究那些在分数上显示出高偏差的较大影响,或持续几个周期的影响。
|
multiqc
FastQ Screen

若有收获,就点个赞吧
0 人点赞
