Introduction to Genome Sequencing**
Recall from “Computing GC Content” that almost all humans share approximately 99.9% of the same nucleotides on the genome. Thus, if we know only a few complete genomes from the species, then we already possess an important key to unlocking the species genome.
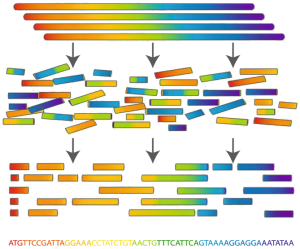
| Determining an organism’s complete genome (called genome sequencing) forms a central task of bioinformatics. Unfortunately, we still don’t possess the microscope technology to zoom into the nucleotide level and determine the sequence of a genome’s nucleotides, one at a time. However, researchers can apply chemical methods to generate and identify much smaller snippets of DNA, called reads. After obtaining a large collection of reads from multiple copies of the same genome, the aim is to reconstruct the desired genome from these small pieces of DNA; this process is called fragment assembly (see Figure 1). |
 Figure 1. Fragment Assembly works by blasting many copies of the same genome into smaller, identifiable reads, which are then used to computationally assemble one copy of the genome. |
|---|---|
Problem
For a collection of strings, a larger string containing every one of the smaller strings as a substring is called a superstring.
By the assumption of parsimony, a shortest possible superstring over a collection of reads serves as a candidate chromosome.
Given: At most 50 DNA strings of approximately equal length, not exceeding 1 kbp, in FASTA format (which represent reads deriving from the same strand of a single linear chromosome).
The dataset is guaranteed to satisfy the following condition: there exists a unique way to reconstruct the entire chromosome from these reads by gluing together pairs of reads that overlap by more than half their length.
Return: A shortest superstring containing all the given strings (thus corresponding to a reconstructed chromosome).
Sample Dataset
Rosalind_56
ATTAGACCTG
>Rosalind_57
CCTGCCGGAA
>Rosalind_58
AGACCTGCCG
>Rosalind_59
GCCGGAATACSample Output
Extra Information
Although the goal of fragment assembly is to produce an entire genome, in practice it is only possible to construct several contiguous portions of each chromosome, called contigs. Furthermore, the assumption made above that reads all derive from the same strand is also practically unrealistic; in reality, researchers will not know the strand of DNA from which a given read has been sequenced.
Solution
主要讲基因组测序后如何组装,组装的过程大致是什么。题目是真实情况的简化,也就是给定数据输出结果一定唯一。但是在实际中多条
reads的重叠部分不可能是唯一的,即不是题中限制多条reads重叠区域大于等于其长度一半。
算法流程:根据不同 reads 的 overlap 程度构建出最终的基因组。
ATTAGACCTGCCGGAATACATTAGACCTG >Rosalind_56CCTGCCGGAA >Rosalind_57AGACCTGCCG >Rosalind_58GCCGGAATAC >Rosalind_59
将上面按照起点进行升序排序就更明白题意要求:
ATTAGACCTGCCGGAATACATTAGACCTG >Rosalind_56AGACCTGCCG >Rosalind_58CCTGCCGGAA >Rosalind_57GCCGGAATAC >Rosalind_59
现在看是否更加清晰呢?但是我们如何构造出最终结果呢?
:::tips
题目限制最终结果是唯一和其只能来源于不同 reads 重叠序列大于等于其自身长度一半的原因?
- 确定唯一性的目的是简化真实的基因组组装,也就是将题目简单化。
如何保证最终结果是唯一的?
- 通过限制不同
reads之间overlap至少为其自身长度一半才可以构建最终基因组:可以减少很多可能。 - 题目输入序列之间不会出现大量
overlap:最终拼接结果中处于两段的reads是确定的,处于中间的reads会与其左侧reads和右侧reads的overlap大于等于其自身长度一半(除此之外不会与其它reads的overlap大于等于其自身长度一半)。这样就能保证最终拼接方向(从哪里延申)和唯一性。 :::
- 通过限制不同
计算不同
reads对之间的overlap长度。例如lcs(56, 57), lcs(56, 58), lcs(56, 59)等。由于题目保证最终答案是唯一的,且由 临近 (上述按起点排序)reads最大overlap 拼接而成。因此我们只要找到起点/终点就可以顺/逆推得到最终答案。- 如何确定起点/终点:首先对每个
reads和其余reads进行最长公共子串处理- 如果该
reads和其它reads最长公共子序列长度大于等于自身长度一半的个数等于**2**,则其是中间reads - 如果该
reads和其它reads最长公共子序列长度大于等于自身长度一半的个数等于**1**,则其是起点或终点。根据最长lcs在该reads起始位置可以判断出是起点(endswith)还是终点(startswith)。
- 如果该
算法流程:
- 求解不同
reads之间lcs的值的所有组合
- 根据
 中
中  的个数确定起点或终点。
的个数确定起点或终点。 - 根据起点/终点进行延申,不同
reads延申条件是 ,依次递推到另一个终点结束。
```python
from typing import List, Tuple
import re
,依次递推到另一个终点结束。
```python
from typing import List, Tuple
import re
class Solution: def extern(self, ri: int, reads: List[str], alignments: List[List[Tuple[int]]], visited: List[bool]) -> str: “””根据当前 alignments[ri] 比对信息,挑选出 overlap >= k / 2 进行向左/右延申””” readI = reads[ri] visited[ri] = True for rj in range(len(alignments[ri])): lcsLen, endi, endj = alignments[ri][rj] if lcsLen * 2 >= len(readI): if not visited[rj]: ret = self.extern(rj, reads, alignments, visited)
# reads[rj] 在 reads[ri] 右侧# reads[ri]: ---*******# reads[rj]: *******---# 我们需要问号部分 *******????????if endi > endj:readI = readI + ret[endj + 1:]# reads[rj] 在 reads[ri] 左侧# reads[ri]: *******---# reads[rj]: ---*******# ????????******* 需要问号部分else:readI = ret[0:-endj] + readIreturn readIdef shortestSuperstring(self, reads: List[str]) -> str:"""根据不同 reads 的 overlap 程度拼接最终基因组"""k = len(reads[0])alignments = [[] for _ in range(len(reads))]for ri in range(len(reads)):for rj in range(len(reads)):if ri == rj:alignments[ri].append((0, -1, -1)) # 添加 seqs[ri] 与 seqs[rj] lcs 比对信息continue# 使用 dp 求解maxLcs, endi, endj = 0, 0, 0m, n = len(seqs[ri]), len(seqs[rj])dp = [[0] * (n + 1) for _ in range(m + 1)]for i in range(1, m + 1):for j in range(1, n + 1):if seqs[ri][i-1] == seqs[rj][j-1]:dp[i][j] = dp[i-1][j-1] + 1if dp[i][j] > maxLcs:maxLcs, endi, endj = dp[i][j], i - 1, j - 1# 添加 seqs[ri] 与 seqs[rj] lcs 比对信息alignments[ri].append((maxLcs, endi, endj))# 2. 寻找起点return self.extern(0, reads, alignments, [False] * len(reads))
fasta = “””
Rosalind_56 ATTAGACCTG Rosalind_57 CCTGCCGGAA Rosalind_58 AGACCTGCCG Rosalind_59 GCCGGAATAC “””
seqs = [mat.group(2).replace(‘\n’, ‘’) for mat in re.finditer(r’>(.)\n((?s:[^>]))’, fasta)] print(Solution().shortestSuperstring(seqs))
另一个算法超时:```pythonfrom typing import List, Tupleimport reclass Solution:def longestCommonSubstr(self, s: str, t: str) -> Tuple[int]:"""求解 s, t 的最长公共子串,返回 (maxLcs, endI, endJ)"""# 使用 dp 求解maxLcs, endi, endj = 0, 0, 0m, n = len(s), len(t)dp = [[0] * (n + 1) for _ in range(m + 1)]for i in range(1, m + 1):for j in range(1, n + 1):if s[i-1] == t[j-1]:dp[i][j] = dp[i-1][j-1] + 1if dp[i][j] > maxLcs:maxLcs, endi, endj = dp[i][j], i - 1, j - 1return (maxLcs, endi, endj)def shortestSuperstring(self, reads: List[str]) -> str:"""根据不同 reads 的 overlap 程度拼接最终基因组"""n = len(reads)k = len(reads[0])genome = reads[0]novisited = [True] * nnovisited[0] = Falsewhile any(novisited):maxLcs, endi, endj, rj = 0, 0, 0, -1for j in range(n):if novisited[j]: # 没被看_maxLcs, _endi, _endj = self.longestCommonSubstr(genome, reads[j])if _maxLcs > maxLcs:maxLcs, endi, endj, rj = _maxLcs, _endi, _endj, jnovisited[rj] = Falseif endj == k - 1: # reads[rj] + genomegenome = reads[rj][:-endj] + genomeelse: # genome ,reads[rj]genome = genome + reads[rj][endj+1:]return genomefasta = """>Rosalind_56ATTAGACCTG>Rosalind_57CCTGCCGGAA>Rosalind_58AGACCTGCCG>Rosalind_59GCCGGAATAC"""seqs = [mat.group(2).replace('\n', '')for mat in re.finditer(r'>(.*)\n((?s:[^>]*))', fasta)]print(Solution().shortestSuperstring(seqs))
复杂度分析:设 reads 数量为 n ,单条 reads 长度为 k , genome 长度为 m (随着合并变长)
- 时间复杂度:
- 最外层
while合并消耗 次;
次; - 内层循环次数与剩余合并数量有关,设最外层循环次数为
i,则内层循环走n - 1 - i次 lcs计算要 ,且随着
,且随着 genome长度变大,即m变大后每次执行lcs时间复杂度变高。- 综上总的时间复杂度为

- 最外层
- 空间复杂度:,求解
lcs问题的创建dp数组。
其实上面算法已经接近正确答案,但是问题出在重复进行多次 lcs,因为若当前 genome 和一个 reads 重叠长度大于等于  就可以进行合并,但是上述算法依旧和其它
就可以进行合并,但是上述算法依旧和其它 reads 继续做 lcs ,最后选择最优进行合并(这种适合在实践中进行,因为本题给的用例比较特殊)。
鉴于每次只需要找到某条 reads 和 genome 的前后缀的重叠长度范围在  即可,因此中间部分的匹配可以省略(尤其是
即可,因此中间部分的匹配可以省略(尤其是 genome 长度不断变长后复杂度剧增时优化非常明显,因为最后  ),则总时间复杂度为
),则总时间复杂度为 
from typing import Listimport reclass Solution:def shortestSuperstring(self, reads: List[str]) -> str:"""根据不同 reads 的 overlap 程度拼接最终基因组"""n = len(reads)genome = reads[0]visited = [False] * nvisited[0] = Truefor _ in range(n - 1):for j in range(n):if not visited[j]: # 没被看visited[j] = Truem = len(reads[j])for length in range(m // 2, m + 1):# genome ****---# reads[j] ---****if genome[:length] == reads[j][-length:]:genome = reads[j][0:-length] + genomebreak# genome ---****# reads[j] ****---if genome[-length:] == reads[j][:length]:genome = genome + reads[j][length:]breakelse: # 无 overlap 候选visited[j] = Falsereturn genomefasta = """>Rosalind_56ATTAGACCTG>Rosalind_57CCTGCCGGAA>Rosalind_58AGACCTGCCG>Rosalind_59GCCGGAATAC"""seqs = list(seq.replace('\n', '') for seq in re.findall('[ATCG\n]+', fasta) if seq.replace('\n', ''))ret = Solution().shortestSuperstring(seqs)print(ret)
复杂度分析:设 reads 数量为 n ,单条 reads 长度为 k , genome 长度为 m (随着合并变长)
- 时间复杂度:
- 最外层
while合并消耗 次; - 内层循环次数与剩余合并数量和当前
genome是否“匹配”一个reads有关,设最外层循环次数为i,最坏情况下内层循环走n - 1 - i次,均摊为 ,因为最坏情况下只可能遇到一次。
,因为最坏情况下只可能遇到一次。 overlap计算要 ,且与
,且与 genome长度无关。- 综上总的时间复杂度为

- 最外层
- 空间复杂度:,不计算切片消耗。

若有收获,就点个赞吧
0 人点赞

{kind=link}