Liu G, Zeng X, Wu B, et al. RNA-Seq analysis of peripheral blood mononuclear cells reveals unique transcriptional signatures associated with radiotherapy response of nasopharyngeal carcinoma and prognosis of head and neck cancer[J]. Cancer biology & therapy, 2020, 21(2): 139-146.
读入表达矩阵
发现上次的一个错误:
Ensembl ID 转换 symbol 后,表达矩阵没有去重,行名有重复(虽然不知道为什么没有报错….
library(AnnotationDbi)library(org.Hs.eg.db)library(survival)library(survminer)
expr <- read.table('../TCGA-HNSC.htseq_counts.tsv',header = T,sep='\t')dim(expr)# [1] 60488 547tail(expr[,1])# [1] "ENSGR0000281849.1" "__no_feature" "__ambiguous"# [4] "__too_low_aQual" "__not_aligned" "__alignment_not_unique"expr <- expr[1:(nrow(expr)-5),]class(expr[,1])# [1] "factor"expr[,1] <- as.character(expr[,1])for (i in 1:nrow(expr)) {expr[i,1] <- strsplit(expr[i,1],"\\.")[[1]][1]}symb <- AnnotationDbi::select(org.Hs.eg.db,expr[,1],"SYMBOL","ENSEMBL")symbtrim <- symb[match(unique(symb$SYMBOL),symb$SYMBOL),] %>% na.omit()row.names(expr) <- expr[,1]expr <- expr[,-1]expr <- expr[symbtrim$ENSEMBL,]row.names(expr) <- symbtrim$SYMBOL
- 更正了之前log方法
- 筛选样本
Sample Type Codes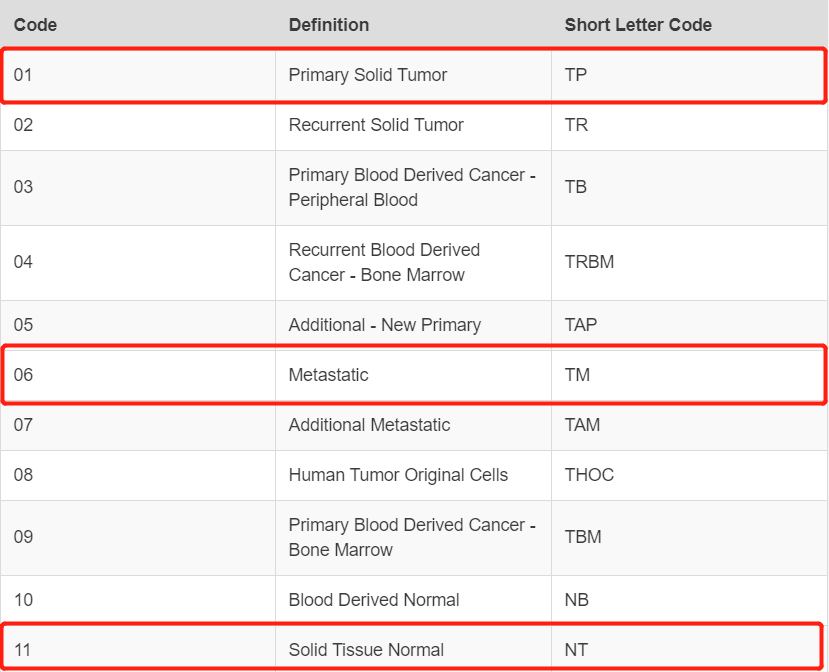
expr <- log2(edgeR::cpm(expr)+1)kp <- substring(colnames(expr),14,15)=='01';table(kp)# kp# FALSE TRUE# 46 500table(substring(colnames(expr),14,15))## 01 06 11# 500 2 44expr <- expr[,kp]dim(expr)# [1] 25483 500save(expr, file = "HNSC_expr.Rdata")
文章中的样本量是497,现在取500
The differential abundance of the expression of radiotherapy-associated genes was further tested in a total of 497 HNSC samples downloaded from TCGA.
读入临床信息
这次发现一个问题

## 上次并没有读取完整clin <- read.table('./TCGA-HNSC.GDC_phenotype.tsv/TCGA-HNSC.GDC_phenotype.tsv',header = T,sep='\t')dim(clin)# [1] 306 139clin <- read.table('./TCGA-HNSC.GDC_phenotype.tsv/TCGA-HNSC.GDC_phenotype.tsv',header = T,sep='\t',fill = T,quote = "")dim(clin)# [1] 612 139
clin$submitter_id.samples = gsub('-','.',clin$submitter_id.samples)row.names(clin) <- clin[,1]phe <- clin[substring(clin$submitter_id.samples,14,15)=='01',]pid <- intersect(colnames(expr),rownames(phe))expr <- expr[,pid]phe <- phe[pid,]phe <- phe[,c("race.demographic","age_at_initial_pathologic_diagnosis","clinical_stage","radiation_therapy")]
surv <- read.table('../TCGA-HNSC.survival.tsv',header = T,sep='\t')surv$sample = gsub('-','.',surv$sample)row.names(surv) <- surv[,1]surv <- surv[pid,]phe <- cbind(phe,surv[,c("OS.time","OS")])colnames(phe) <- c("race","age","stage","radiation","time","event")save(phe, file = "HNSC_phe.Rdata")
Cox regression
signature <- expr[c("CHAC2","CLEC9A","GNG10","JCHAIN","KLRB1","NOG","OLR1","PRELID2","SYT1","VWCE","ZNF443"),]mySurv=with(phe,Surv(time, event))
univariate cox regression
unicox <-apply(signature, 1 ,function(gene){group=ifelse(gene>median(gene),'high','low')survival_dat <- data.frame(group=group,stage=phe$stage,age=phe$age,race=phe$race,radiation=phe$radiation,stringsAsFactors = F)m=coxph(mySurv ~group, data = survival_dat)beta <- coef(m)se <- sqrt(diag(vcov(m)))HR <- exp(beta)HRse <- HR * setmp <- round(cbind(coef = beta, se = se, z = beta/se, p = 1 - pchisq((beta/se)^2, 1),HR = HR, HRse = HRse,HRz = (HR - 1) / HRse, HRp = 1 - pchisq(((HR - 1)/HRse)^2, 1),HRCILL = exp(beta - qnorm(.975, 0, 1) * se),HRCIUL = exp(beta + qnorm(.975, 0, 1) * se)), 3)return(tmp['grouplow',])})unicox=t(unicox)table(unicox[,4]<0.05)## FALSE TRUE# 3 8
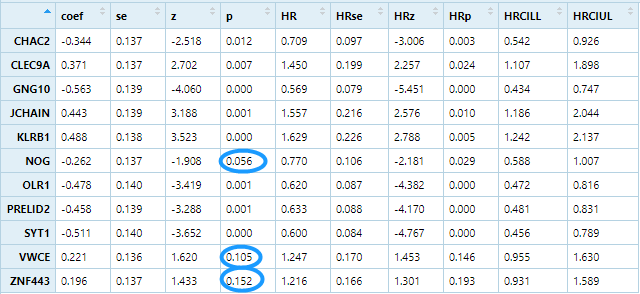
multivariate cox regression
multicox <-apply(signature, 1 ,function(gene){group=ifelse(gene>median(gene),'high','low')survival_dat <- data.frame(group=group,stage=phe$stage,age=phe$age,race=phe$race,radiation=phe$radiation,stringsAsFactors = F)m=coxph(mySurv ~ age + stage+ group+radiation+race, data = survival_dat)beta <- coef(m)se <- sqrt(diag(vcov(m)))HR <- exp(beta)HRse <- HR * setmp <- round(cbind(coef = beta, se = se, z = beta/se, p = 1 - pchisq((beta/se)^2, 1),HR = HR, HRse = HRse,HRz = (HR - 1) / HRse, HRp = 1 - pchisq(((HR - 1)/HRse)^2, 1),HRCILL = exp(beta - qnorm(.975, 0, 1) * se),HRCIUL = exp(beta + qnorm(.975, 0, 1) * se)), 3)return(tmp['grouplow',])})multicox=t(multicox)table(multicox[,4]<0.05)## FALSE TRUE# 2 9
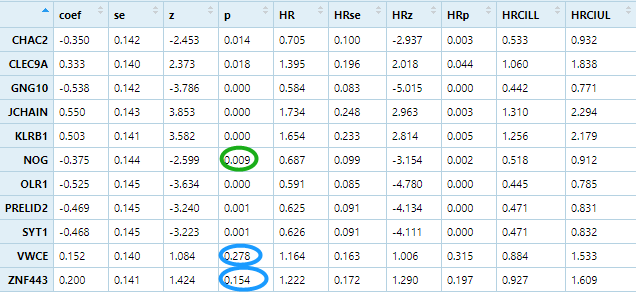
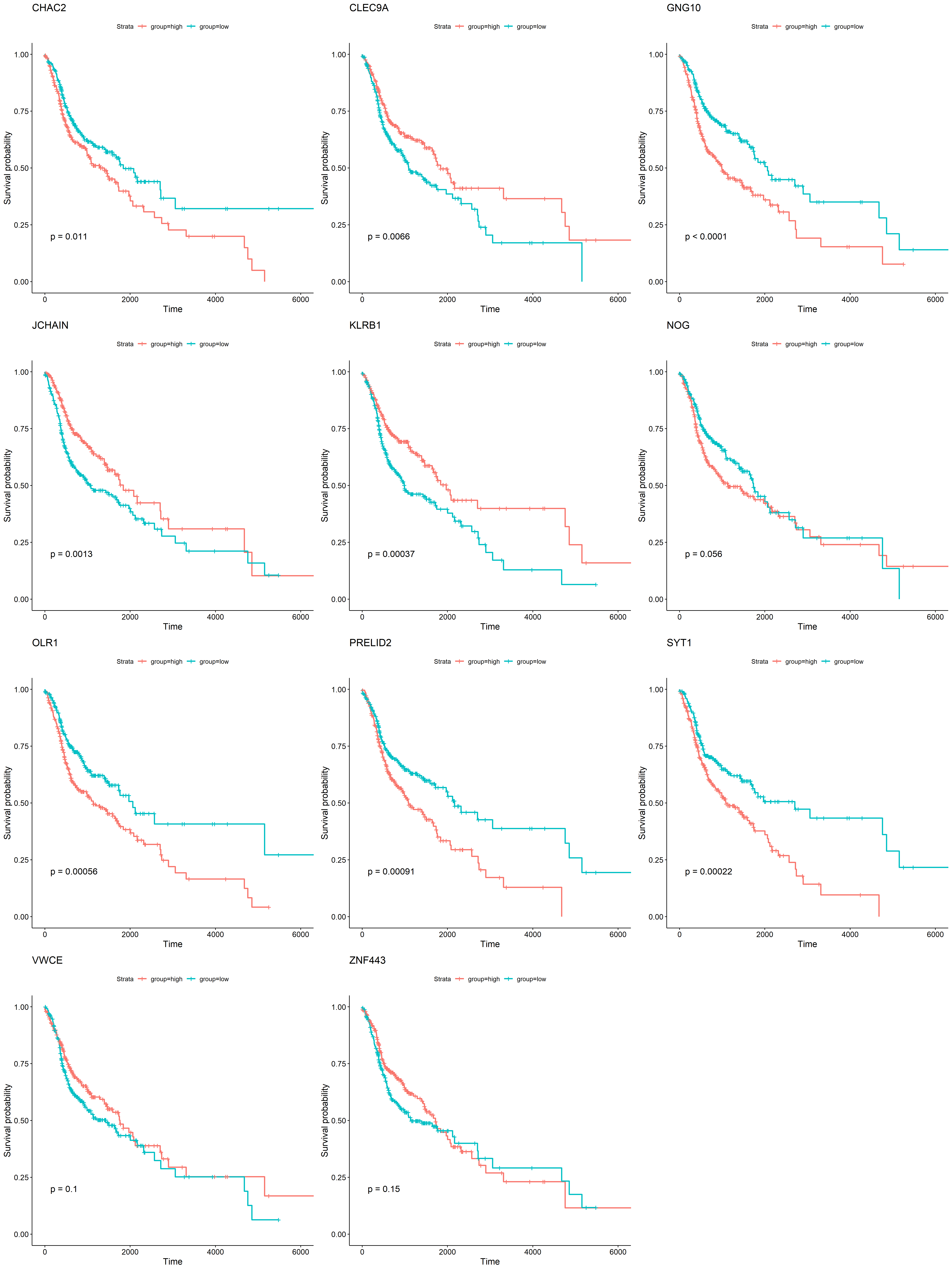
KM Plot
for (i in 1:nrow(signature)) {gene=as.numeric(signature[i,])phe$group=ifelse(gene>median(gene),'high','low')fit <- survfit(Surv(time, event) ~ group,data = phe)survp=ggsurvplot(fit,pval = TRUE,title=row.names(signature)[i])ggsave(filename = paste0('TCGA_gene_',i,'.png'),print(survp))}

若有收获,就点个赞吧
0 人点赞
