陷入怀疑😓
signature 生存分析复现
Xu G, Zhang M, Zhu H, et al. A 15-gene signature for prediction of colon cancer recurrence and prognosis based on SVM[J]. Gene, 2017, 604: 33-40.
identify signature
library(GEOquery)library(AnnotationDbi)library(org.Hs.eg.db)
DEG
- GSE17537(以这个数据集为基础找差异表达基因)
data in GSE17537 were analyzed using the Linear Models for Microarray data (LIMMA) method to identify the differentially expressed genes (DEGs). Preprocessed data were normalized by Z-score transformation… The threshold for identification of DEGs was set as p < 0.05 and |logFC(fold change)| > 0.7.
f <- 'GSE17537.Rdata'if(!file.exists(f)){gset <- getGEO('GSE17537',destdir = '.',AnnotGPL = F,getGPL = F)save(gset,file=f)}load('GSE17537.Rdata')degexpr <- exprs(gset[[1]])degpdat <- pData(gset[[1]])degexpr[1:4,1:4]# GSM437270 GSM437271 GSM437272 GSM437273# 1007_s_at 10.636083 11.042240 11.015748 10.734469# 1053_at 7.974253 8.542731 7.190542 8.731533# 117_at 8.242136 7.271520 7.368352 7.027436# 121_at 9.315796 9.277670 9.325623 9.398863## z-scoredegexpr <- t(scale(t(degexpr)))degexpr[1:4,1:4]# GSM437270 GSM437271 GSM437272 GSM437273# 1007_s_at -0.6468308 0.4474904 0.3761127 -0.3817476# 1053_at -0.6561249 0.3432638 -2.0338972 0.6751799# 117_at 1.8993885 0.1023676 0.2816449 -0.3495356# 121_at -1.0241496 -1.2322858 -0.9705000 -0.5706729degsymb <- select(hgu133plus2.db,row.names(degexpr),"SYMBOL","PROBEID")degtrim <- degsymb[match(unique(degsymb$SYMBOL),degsymb$SYMBOL),]degtrim <- na.omit(degtrim)degexpr <- degexpr[degtrim$PROBEID,]row.names(degexpr) <- degtrim$SYMBOLdim(degexpr)# [1] 1399 55save(degexpr, file = "GEO-degexpr.Rdata")save(degpdat,file = "GEO-degpdat.Rdata")
DEG筛选rm(list = ls())load("GEO-degexpr.Rdata")load("GEO-degpdat.Rdata")library(limma)table(degpdat$`dfs_event (disease free survival; cancer recurrence):ch1`)## no recurrence recurrence# 36 19group_list <- degpdat$`dfs_event (disease free survival; cancer recurrence):ch1`design <- model.matrix(~0+factor(group_list))colnames(design) <- c("no", "re")rownames(design) <- colnames(degexpr)contrast.matrix <- makeContrasts('no-re',levels = design)fit <- lmFit(degexpr, design)fit1 <- contrasts.fit(fit, contrast.matrix)fit2 <- eBayes(fit1)DEGstb <- topTable(fit2, coef=1, n=Inf)DEGmtx <- na.omit(DEGstb)DEGmtx <- DEGmtx[absDEGmtx$logFC]DEGmtx$result <- ifelse(DEGmtx$P.Value < 0.05 &abs(DEGmtx$logFC) > 0.7,ifelse(DEGmtx$logFC > 0.7,"UP", "DOWN"), "NOT")table(DEGmtx$result)## DOWN NOT UP# 14 1314 71head(DEGmtx)# logFC AveExpr t P.Value adj.P.Val B result# DDX31 1.1059228 2.267284e-15 3.931463 8.450714e-05 0.1182255 0.8853818 UP# C11orf42 0.9940823 -5.083371e-16 3.533879 4.097569e-04 0.1661362 -0.3317710 UP# ACACB 0.9625943 3.113513e-15 3.421942 6.220910e-04 0.1661362 -0.6510647 UP# PGLS 0.9068060 -8.080973e-16 3.223619 1.266363e-03 0.1661362 -1.1915345 UP# STARD6 0.9032174 -1.506371e-16 3.210862 1.323938e-03 0.1661362 -1.2251955 UP# IRAK2 0.8952166 4.313153e-16 3.182420 1.461101e-03 0.1661362 -1.2997641 UP
文献中的DEG筛选结果:A total of 1207 genes were identified as DEGs between recurrence and no-recurrence samples, including 726 downregulated and 481 upregulated genes.
我的筛选结果:table(DEGmtx$result)## DOWN NOT UP# 14 1314 71save(DEGmtx, file = "DEGs.Rdata")
signature
To obtain the optimal feature genes for diagnosing recurrence, SVM-RFE algorithm was performed using genes as features and their expression levels as feature values. As a result, 85% prediction accuracy was found to be achieved using a 15-gene combination.
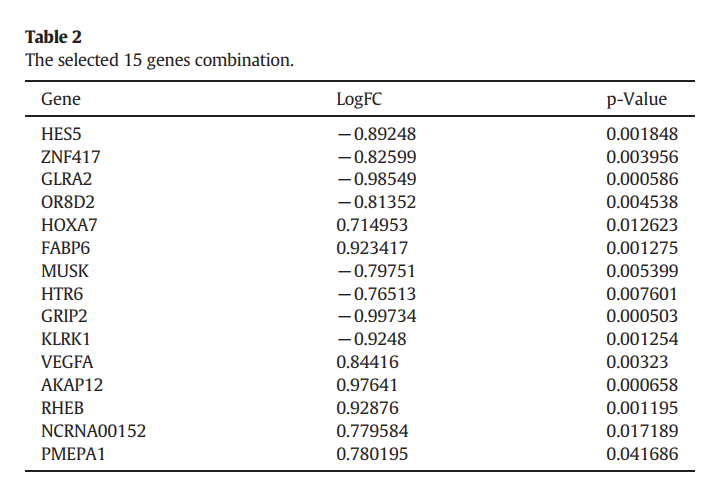
这一步应该在我浅薄的能力之外…
直接在前面得到的DEG中找这15个基因:
paper_signature <- DEGmtx[c("HES5","ZNF417","GLRA2","OR8D2","HOXA7","FABP6","MUSK","HTR6","GRIP2","KLRK1","VEGFA","AKAP12","RHEB","NCRNA00152","PMEPA1"),]
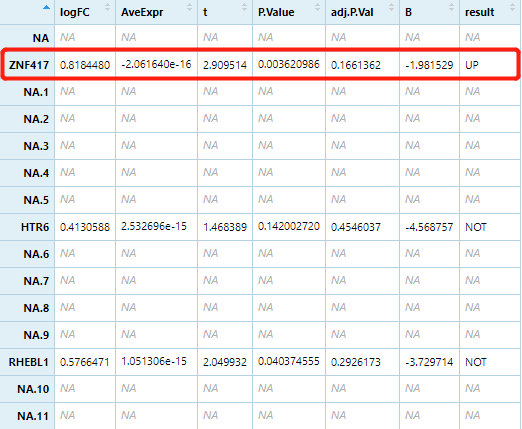
但只有3个基因在筛选的DEG/下载的表达矩阵中,只有 ZNF417 为差异表达基因。
鉴于并不知道怎么得到signature….依然只能直接进行文章中的 Validation analysis
数据准备
GEO
- GSE38832
if(!file.exists(f)){gset <- getGEO('GSE38832',destdir = '.',AnnotGPL = F,getGPL = F)save(gset,file=f)}load('GSE38832.Rdata')expr1 <- exprs(gset[[1]])pdat1 <- pData(gset[[1]])expr1[1:4,1:4]# GSM950411 GSM950412 GSM950413 GSM950414# 1007_s_at 12.015001 12.319230 12.268841 12.309278# 1053_at 9.600642 9.991989 9.349406 8.934902# 117_at 8.125449 7.661441 11.611829 8.575235# 121_at 10.868107 11.191637 10.794642 11.157243expr1 <- log2(expr1+1)symb1 <- select(hgu133plus2.db,row.names(expr1),"SYMBOL","PROBEID")trim1 <- symb1[match(unique(symb1$SYMBOL),symb1$SYMBOL),]trim1 <- na.omit(trim1)expr1 <- expr1[trim1$PROBEID,]row.names(expr1) <- trim1$SYMBOLsave(expr1, file = "GEO-expr1.Rdata")
- GSE17538
只有6个样本,并非文献中的200多个,ID转换过程中发现下载到的是小鼠…?
去官网直接下载,发现好像失效了,无法下载——弃。

f <- 'GSE17538.Rdata'if(!file.exists(f)){gset <- getGEO('GSE17538',destdir = '.',AnnotGPL = F,getGPL = F)save(gset,file=f)}load('GSE17538.Rdata')expr2 <- exprs(gset[[1]])pdat2 <- pData(gset[[1]])expr2[1:4,1:4]# GSM472198 GSM472199 GSM472200 GSM472201# 1415670_at 9.284539 9.239952 9.311207 9.501534# 1415671_at 10.668639 10.677101 10.637565 10.872466# 1415672_at 10.624298 10.563723 10.540626 10.685249# 1415673_at 10.323815 10.267230 10.453991 9.791176expr2 <- log2(expr2+1)BiocManager::install("mouse4302.db")library(mouse4302.db)symb2 <- select(mouse4302.db,row.names(expr2),"SYMBOL","PROBEID")trim2 <- symb2[match(unique(symb2$SYMBOL),symb2$SYMBOL),]trim2 <- na.omit(trim2)expr2 <- expr2[trim2$PROBEID,]row.names(expr2) <- trim2$SYMBOLsave(expr2, file = "GEO-expr2.Rdata")
- GSE28814

没有找到对应的注释R包,从 GPL10379 Rosetta/Merck Human RSTA Custom Affymetrix 2.0 microarray [HuRSTA-2a520709] Data table 手动导出txt。
需要在excel中打开调整分隔符重新保存,才能在R中正确读取。
f <- 'GSE28814.Rdata'if(!file.exists(f)){gset <- getGEO('GSE28814',destdir = '.',AnnotGPL = F,getGPL = F)save(gset,file=f)}load('GSE28814.Rdata')expr3 <- exprs(gset[[1]])pdat3 <- pData(gset[[1]])expr3[1:4,1:4]# GSM707237 GSM707238 GSM707239 GSM707240# 100121619_TGI_at 2.4105105 1.9840042 2.9346733 2.4020889# 100121620_TGI_at 2.3519606 2.1129721 2.0588146 1.6712892# 100121621_TGI_at 1.2706607 0.7851753 1.4145823 1.5996047# 100121622_TGI_at 0.8892922 1.0277808 0.6097267 0.7636354anno <- read.table('./HuRSTA-2a520709.txt',sep = "\t",header = T,na.strings = NA, stringsAsFactors = F)anno[1:4,1:4]# ID EntrezGeneID GeneSymbol GB_ACC# 1 100138926_TGI_at 6566 SLC16A1 BC026317# 2 100131045_TGI_at NA BX098869# 3 100134882_TGI_at 3458 IFNG NM_000619# 4 100149534_TGI_at 51552 RAB14 AK090889
TCGA
- GDC TCGA Colon Cancer (COAD) TCGA Colon Cancer (COAD)&removeHub=https%3A%2F%2Fxena.treehouse.gi.ucsc.edu%3A443) (15 datasets)
- HTSeq - Counts (n=512) GDC Hub
- Phenotype (n=571) GDC Hub
- survival data (n=539) GDC Hub
expr4 <- read.table('./TCGA-COAD.htseq_counts.tsv/TCGA-COAD.htseq_counts.tsv',header = T,sep='\t')tail(expr4[,1])# [1] ENSGR0000281849.1 __no_feature __ambiguous# [4] __too_low_aQual __not_aligned __alignment_not_unique# 60488 Levels: __alignment_not_unique __ambiguous __no_feature __not_aligned ... ENSGR0000281849.1class(expr4[,1])# [1] "factor"expr4[,1] <- as.character(expr4[,1])for (i in 1:nrow(expr4)) {expr4[i,1] <- strsplit(expr4[i,1],"\\.")[[1]][1]}row.names(expr4) <- expr4[,1]expr4 <- expr4[,-1]symb4 <- select(org.Hs.eg.db,expr4[,1],"SYMBOL","ENSEMBL")trim4 <- symb4[match(unique(symb4$SYMBOL),symb4$SYMBOL),]trim4 <- na.omit(trim4)expr4 <- expr4[trim4$ENSEMBL,]row.names(expr4) <- trim4$SYMBOLexpr4[1:4,1:4]# TCGA.DM.A288.01A TCGA.QL.A97D.01A TCGA.CM.6164.01A TCGA.G4.6299.01A# TSPAN6 10.985842 12.391512 13.857787 9.807355# TNMD 3.700440 7.531381 6.714246 1.000000# DPM1 11.163650 11.798472 12.496354 11.004220# SCYL3 9.262095 9.581201 9.419960 9.903882expr4 <- log2(expr4+1)save(expr4, file = "TCGA-expr.Rdata")
Validation analysis
- GSE38832
vali1 <- expr1[c("HES5","ZNF417","GLRA2","OR8D2","HOXA7","FABP6","MUSK","HTR6","GRIP2","KLRK1","VEGFA","AKAP12","RHEB","NCRNA00152","PMEPA1"),]

- GSE28814
vali3 <- expr3[c("HES5","ZNF417","GLRA2","OR8D2","HOXA7","FABP6","MUSK","HTR6","GRIP2","KLRK1","VEGFA","AKAP12","RHEB","NCRNA00152","PMEPA1"),]

结果居然一个都没有….
- TCGA
vali4 <- expr4[c("HES5","ZNF417","GLRA2","OR8D2","HOXA7","FABP6","MUSK","HTR6","GRIP2","KLRK1","VEGFA","AKAP12","RHEB","NCRNA00152","PMEPA1"),]row.names(vali4)# [1] "HES5" "ZNF417" "GLRA2" "OR8D2" "HOXA7" "FABP6" "MUSK" "HTR6" "GRIP2"# [10] "KLRK1" "VEGFA" "AKAP12" "RHEB" "NA" "PMEPA1"
缺少 NCRNA00152
KM plot (TCGA data)
vali4 <- expr4[c("HES5","ZNF417","GLRA2","OR8D2","HOXA7","FABP6","MUSK","HTR6","GRIP2","KLRK1","VEGFA","AKAP12","RHEB","NCRNA00152","PMEPA1"),]row.names(vali4)vali4 <- na.omit(vali4)clin$submitter_id.samples = gsub('-','.',clin$submitter_id.samples)row.names(clin) <- clin[,1]#### 筛选复发病人,并无有用信息table(clin$progression_or_recurrence.diagnoses)## not reported# 2 268surv$sample <- gsub('-','.',surv$sample)row.names(surv) <- surv[,1]surv <- surv[surv$sample %in% row.names(clin),]clin <- clin[row.names(clin) %in% surv$sample,]phe <- cbind(clin,surv[,c("OS.time","OS")])vali4 <- vali4[,colnames(vali4) %in% row.names(phe)]phe <- phe[row.names(phe) %in% colnames(vali4),]dim(phe)vali4 <- as.matrix(vali4)#### 将14个基因视为一个对象for(i in 1:ncol(vali4)){sig[i] <- sum(vali4[,i])}group <- ifelse(sig > median(sig),'high','low')phe$group <- groupphe <- phe[,c("OS.time","OS","group")]ggsurvplot(survfit(Surv(OS.time, OS)~group, data=phe), pval=TRUE)
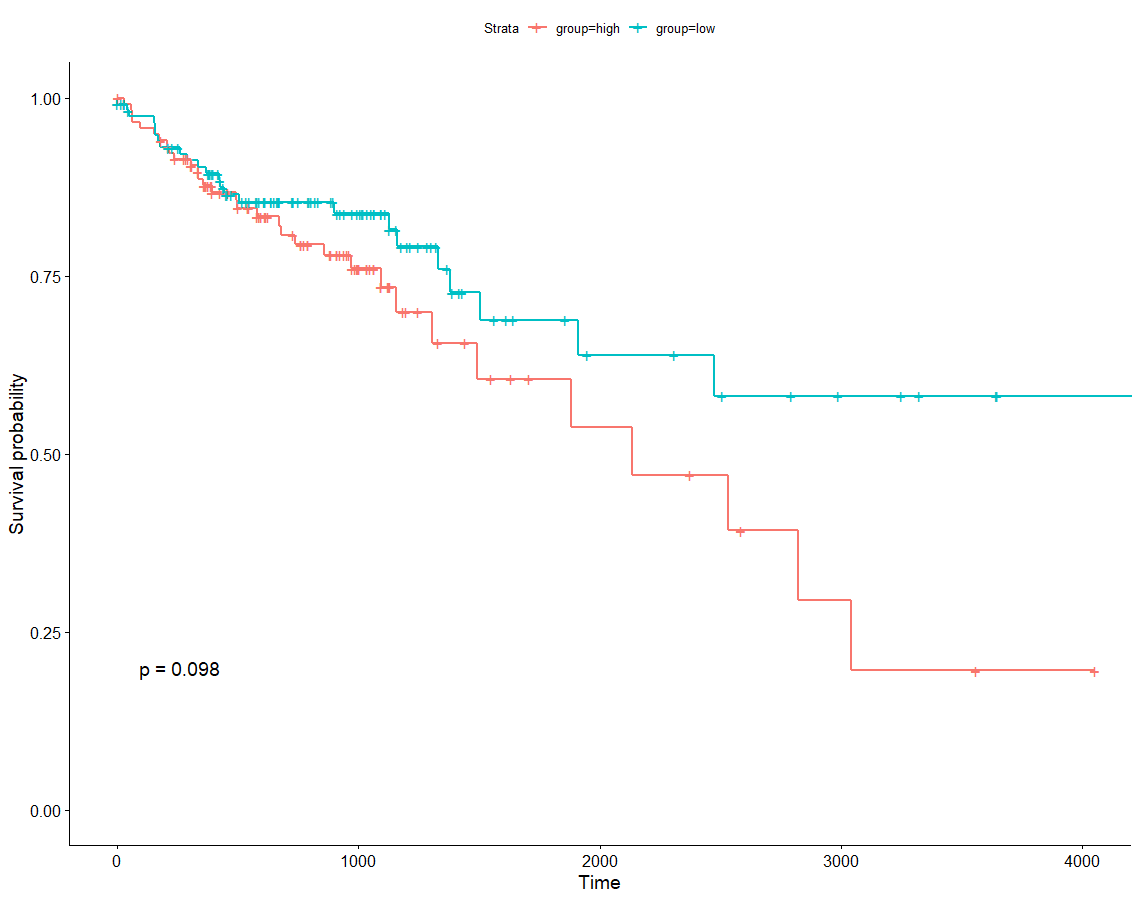
文章中是这样的:
The results indicated that patients with different recurrent risks could exhibit a significantly different
prognosis in two datasets: GSE38832, p = 0.04, Fig. 5A; GSE28814, p = 0.0578, Fig. 5B; and TCGA, p = 0.0162, Fig. 5C.

若有收获,就点个赞吧
0 人点赞
