YTHDF1在卵巢癌的预后
1. 数据准备
下载
[cohort: GDC TCGA Ovarian Cancer (OV)]
- HTSeq - Counts (n=379) GDC Hub
- Phenotype (n=758) GDC Hub
- survival data (n=731) GDC Hub
读入/整合
library(AnnotationDbi)library(org.Hs.eg.db)library(survival)library(survminer)
- 表达矩阵
expr <- read.table('./TCGA-OV.htseq_counts.tsv/TCGA-OV.htseq_counts.tsv',header = T,sep='\t')dim(expr)# [1] 60488 380expr[1:4,1:4]# Ensembl_ID TCGA.23.1120.01A TCGA.29.1695.01A TCGA.61.2003.01A# 1 ENSG00000000003.13 12.308908 12.298063 11.34430# 2 ENSG00000000005.5 3.000000 3.906891 1.00000# 3 ENSG00000000419.11 11.587309 11.433585 11.84549# 4 ENSG00000000457.12 9.147205 10.601771 10.23122tail(expr[,1])# [1] "ENSGR0000281849.1" "__no_feature" "__ambiguous"# [4] "__too_low_aQual" "__not_aligned" "__alignment_not_unique"expr <- expr[1:(nrow(expr)-5),]class(expr[,1])# [1] "factor"expr[,1] <- as.character(expr[,1])for (i in 1:nrow(expr)) {expr[i,1] <- strsplit(expr[i,1],"\\.")[[1]][1]}symb <- select(org.Hs.eg.db,expr[,1],"SYMBOL","ENSEMBL")table(symb[,2]=="YTHDF1")## FALSE TRUE# 25590 1which(symb$SYMBOL=="YTHDF1")# [1] 9300thegene <- symb[9300,]thegene <- expr[expr$Ensembl_ID==thegene$ENSEMBL,]thegene <- thegene[,-1]row.names(thegene) <- "YTHDF1"thegene <- round(thegene,digits = 2)save(thegene,file='YTHDF1expression.Rdata')
- 临床数据
clin <- read.table('./TCGA-HNSC.GDC_phenotype.tsv',header = T,sep='\t')summary(clin)clin[1:4,1:4]clin$submitter_id.samples = gsub('-','.',clin$submitter_id.samples)row.names(clin) <- clin[,1]clin <- clin[c("age_at_initial_pathologic_diagnosis","clinical_stage","person_neoplasm_cancer_status","radiation_therapy","race.demographic")]colnames(clin) <- c("age","stage","status","radiation","race")clin <- na.omit(clin)save(clin,file='clinical.Rdata')
- 生存数据
surv <- read.table('./TCGA-OV.survival.tsv/TCGA-OV.survival.tsv',header = T,sep='\t')
- 整合数据(挑选病人)
surv$sample <- gsub('-','.',surv$sample)row.names(surv) <- surv[,1]surv <- surv[surv$sample %in% row.names(clin),]clin <- clin[row.names(clin) %in% surv$sample,]phe <- cbind(clin,surv[,c("OS.time","OS")])thegene <- thegene[,colnames(thegene) %in% row.names(phe)]phe <- phe[row.names(phe) %in% colnames(thegene),]# table(row.names(phe) %in% colnames(thegene))dim(phe)# [1] 188 7save(phe,file='phe_surv.Rdata')
2. cox
mySurv=with(phe,Surv(OS.time, OS))
univariate cox regression
unicox <-apply(thegene, 1 ,function(gene){group=ifelse(gene>median(gene),'high','low')survival_dat <- data.frame(group=group,stage=phe$stage,age=phe$age,status=phe$status,radiation=phe$radiation,race=phe$race,stringsAsFactors = F)m=coxph(mySurv ~group, data = survival_dat)beta <- coef(m)se <- sqrt(diag(vcov(m)))HR <- exp(beta)HRse <- HR * setmp <- round(cbind(coef = beta, se = se, z = beta/se, p = 1 - pchisq((beta/se)^2, 1),HR = HR, HRse = HRse,HRz = (HR - 1) / HRse, HRp = 1 - pchisq(((HR - 1)/HRse)^2, 1),HRCILL = exp(beta - qnorm(.975, 0, 1) * se),HRCIUL = exp(beta + qnorm(.975, 0, 1) * se)), 3)return(tmp['grouplow',])})unicox=t(unicox)table(unicox[,4]<0.05)## FALSE# 1unicox[,4]# [1] 0.933
multivariate cox regression
multicox <-apply(thegene, 1 ,function(gene){group=ifelse(gene>median(gene),'high','low')survival_dat <- data.frame(group=group,stage=phe$stage,age=phe$age,status=phe$status,radiation=phe$radiation,race=phe$race,stringsAsFactors = F)m=coxph(mySurv ~ age + stage+ group+status+radiation+race, data = survival_dat)beta <- coef(m)se <- sqrt(diag(vcov(m)))HR <- exp(beta)HRse <- HR * setmp <- round(cbind(coef = beta, se = se, z = beta/se, p = 1 - pchisq((beta/se)^2, 1),HR = HR, HRse = HRse,HRz = (HR - 1) / HRse, HRp = 1 - pchisq(((HR - 1)/HRse)^2, 1),HRCILL = exp(beta - qnorm(.975, 0, 1) * se),HRCIUL = exp(beta + qnorm(.975, 0, 1) * se)), 3)return(tmp['grouplow',])})multicox=t(multicox)table(multicox[,4]<0.05)## FALSE# 1multicox[,4]# [1] 0.982
作图
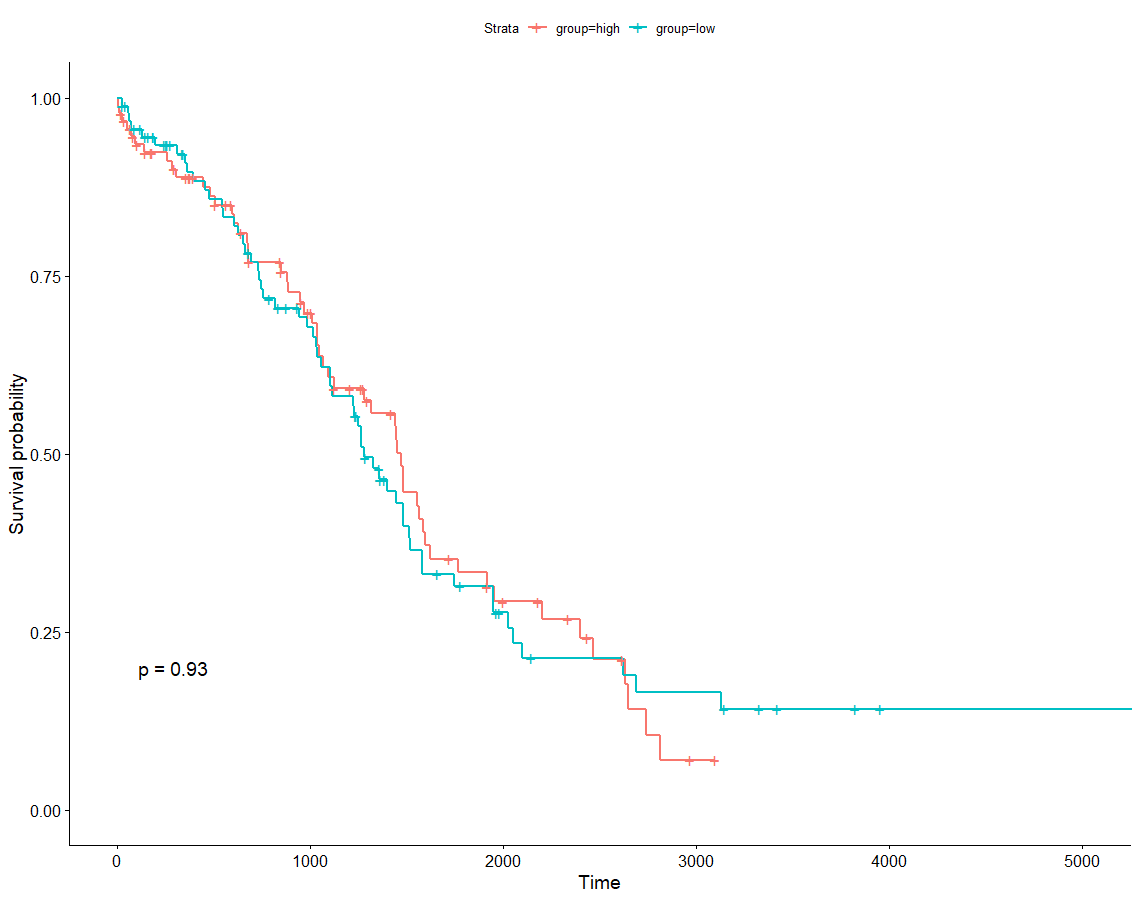
group=ifelse(as.numeric(thegene)>median(as.numeric(thegene)),'high','low')phe$group <- groupggsurvplot(survfit(Surv(OS.time, OS)~group, data=phe), pval=TRUE)

若有收获,就点个赞吧
0 人点赞
