文章:
Cell. 2018 May : Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing.
>
从MSigDB数据库得到6个基因集
下载METABRIC表达矩阵
Breast Cancer (METABRIC, Nature 2012 & Nat Commun 2016)
导出到Motrix里可以以2M/S的速度解决墙内下载问题。
1 读入 data_clinical_patient.txt
clin <- read.table('./brca_metabric/data_clinical_patient.txt',header = T,sep='\t')summary(clin)save(clin,file='metabric_clinical.Rdata')
2 读入 data_expression_median.txt
expr <- read.table('./brca_metabric/data_expression_median.txt',header = T,sep='\t')dim(expr)expr[1:4,1:4]head(colnames(expr),10)length(unique(expr$Hugo_Symbol))gs <- expr$Hugo_Symbolexpr <- expr[,-c(1,2)]
取小数点前两位
expr <- apply(expr, 2, function(x){as.numeric(format(x,digits = 2))})
行名重命名及去除NA值
rownames(expr) <- gsexpr <- na.omit(expr)save(expr,file='metabric_expression.Rdata')
将’-‘替换为’.’,与表达矩阵的列名一致
clin$PATIENT_ID = gsub('-','.',clin$PATIENT_ID)phe <- clin[match(colnames(expr),clin$PATIENT_ID),]
3 GSVA
library(ggplot2)library(clusterProfiler)library(org.Hs.eg.db)library(GSVA)library(GSEABase)library(dplyr)rm(list = ls())d <- './gmt/'gmts <- list.files(d)gmtfile <- lapply(file.path(d,gmts),getGmt)
st <- Sys.time()for(i in 1:length(gmtfile)){assign(paste0("es_max_", i), gsva(expr,gmtfile[[i]],mx.diff=FALSE, verbose=FALSE,parallel.sz=1))}et <- Sys.time()et - st# Time difference of 7.654427 hourses_max <- rbind(es_max_1,es_max_2,es_max_3,es_max_4,es_max_5,es_max_6,es_max_7,es_max_8,es_max_9,es_max_10,es_max_11,es_max_12,es_max_13)es_max[1:4,1:4]save(es_max,file = "GSVA_es_max.Rdata")
4 Survival Analysis
A. 选病人(根据是否化疗)
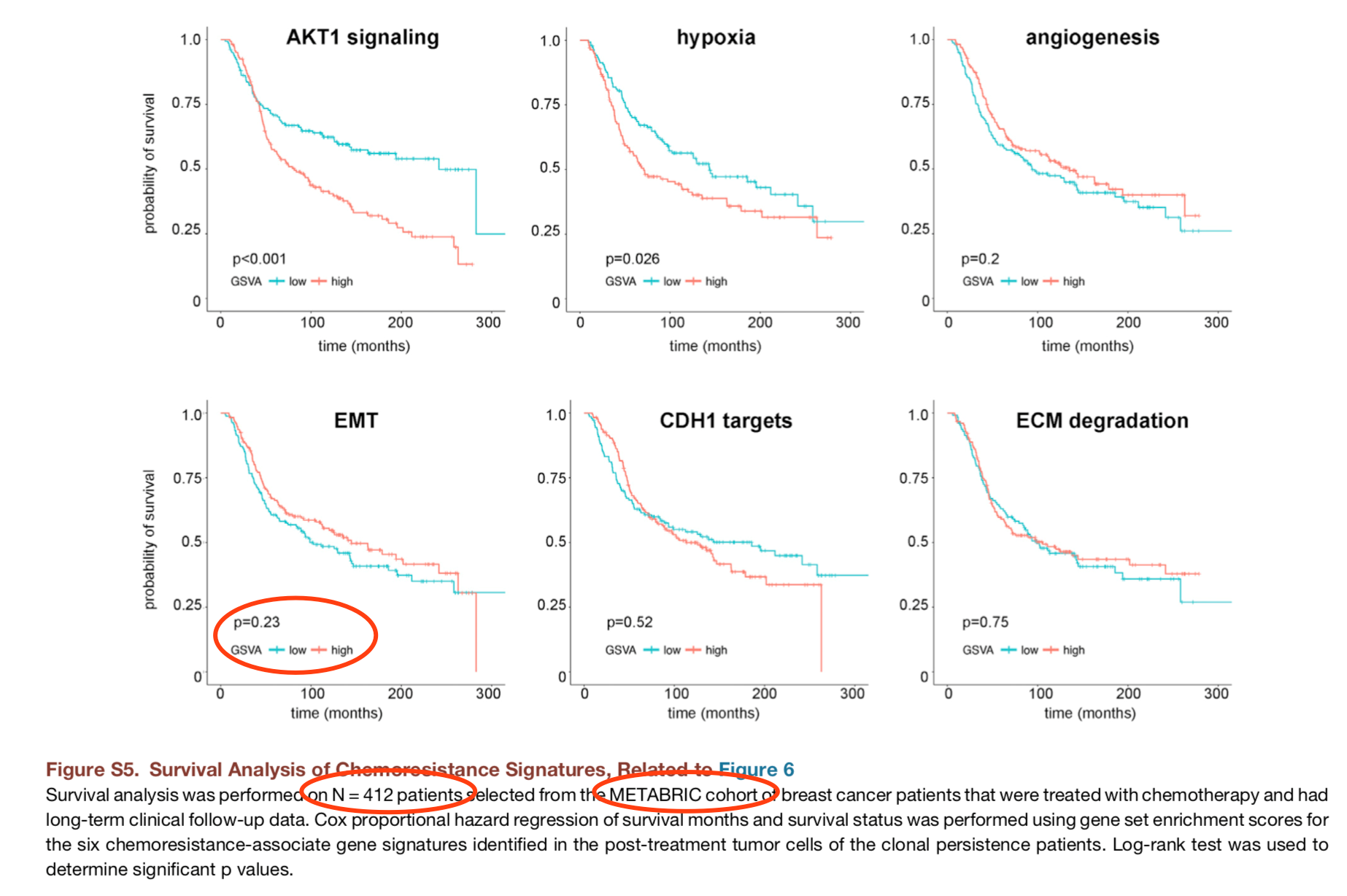
Next, we used an extended cohort of breast cancer patients with chemotherapy (n = 412) from the METABRIC cohort (Curtis et al., 201230365-9?_returnURL=https%3A%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS0092867418303659%3Fshowall%3Dtrue#), Pereira et al., 201630365-9?_returnURL=https%3A%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS0092867418303659%3Fshowall%3Dtrue#)) with gene expression data and long-term clinical follow-up data to determine if any of the chemoresistance-associated signatures correlated with patient survival.
table(clin$CHEMOTHERAPY)patients_selected<- clin[clin$CHEMOTHERAPY=="YES",]dat <- t(es_max)patients_selected$PATIENT_ID = gsub('-','.',patients_selected$PATIENT_ID)row.names(patients_selected) <- patients_selected$PATIENT_IDOSinfo <- patients_selected[,c("OS_MONTHS","OS_STATUS")]dat2 <- dat[row.names(dat) %in% row.names(OSinfo),] %>% na.omit()OSinfo <- OSinfo[row.names(dat2),]
B. 选病人(根据 claudin subtype)
table(clin$CLAUDIN_SUBTYPE)patients_selected_1 <- filter(clin,CLAUDIN_SUBTYPE == "Basal")patients_selected_2 <- filter(clin,CLAUDIN_SUBTYPE == "claudin-low")patients_selected_claudin <- rbind(patients_selected_1, patients_selected_2)dat <- t(es_max)patients_selected_claudin$PATIENT_ID <- gsub('-','.',patients_selected_claudin$PATIENT_ID)row.names(patients_selected_claudin) <- patients_selected_claudin$PATIENT_IDOSinfo <- patients_selected_claudin[,c("OS_MONTHS","OS_STATUS")]dat2 <- dat[row.names(dat) %in% row.names(OSinfo),] %>% na.omit()OSinfo <- OSinfo[row.names(dat2),]
合并分组信息
dat2 <- cbind(dat2,OSinfo)library(reshape2)dat3 <- melt(dat2,c("OS_MONTHS","OS_STATUS"))dat3$OS_STATUS <- ifelse(dat3$OS_STATUS=="DECEASED", 0, 1)colnames(dat3)[3] <- "geneset"save(dat3,file = "df_survivalplot_chem.Rdata")## save(dat3,file = "df_survivalplot_claudin.Rdata")
作图
各个基因集分别作图
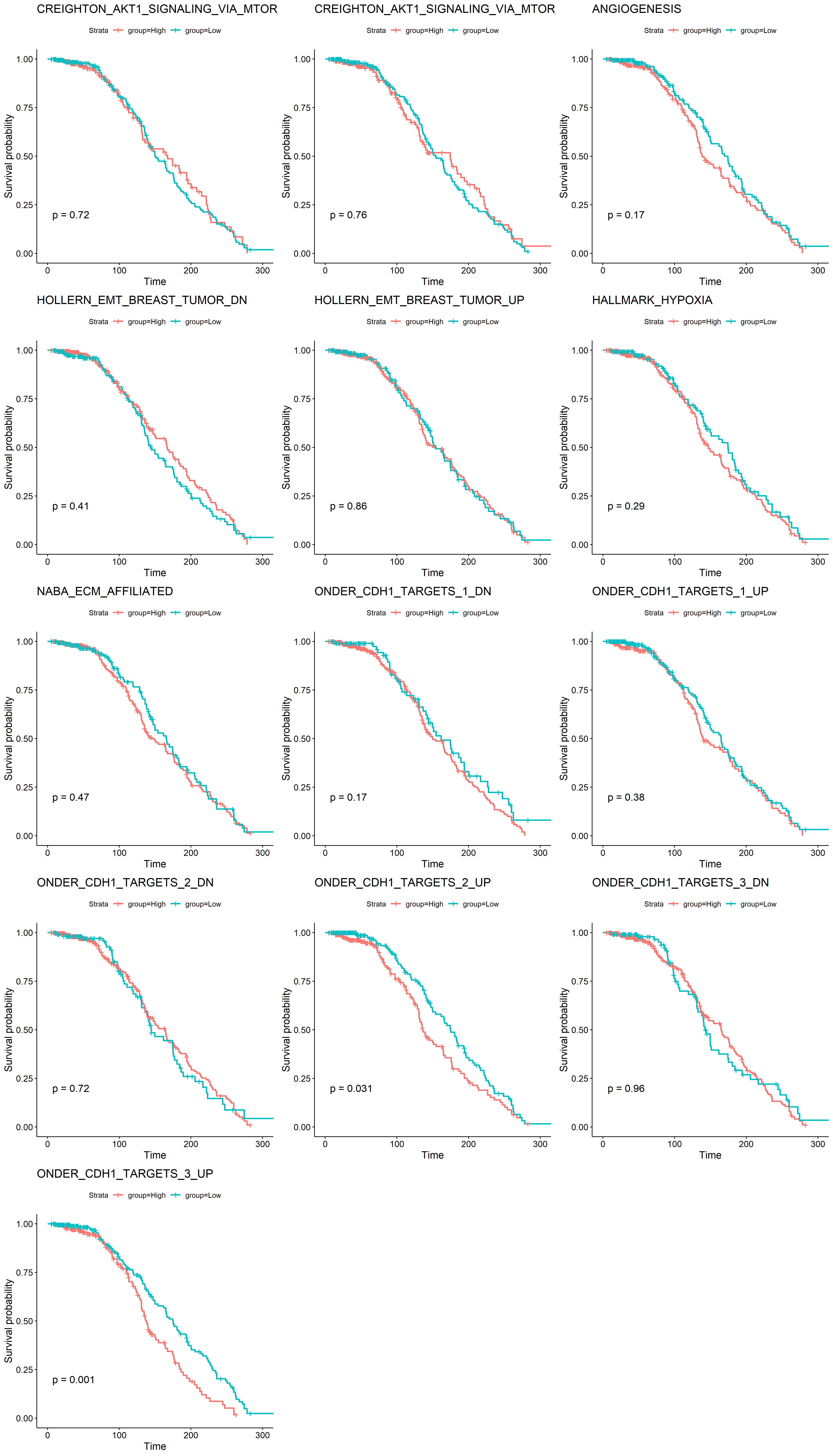
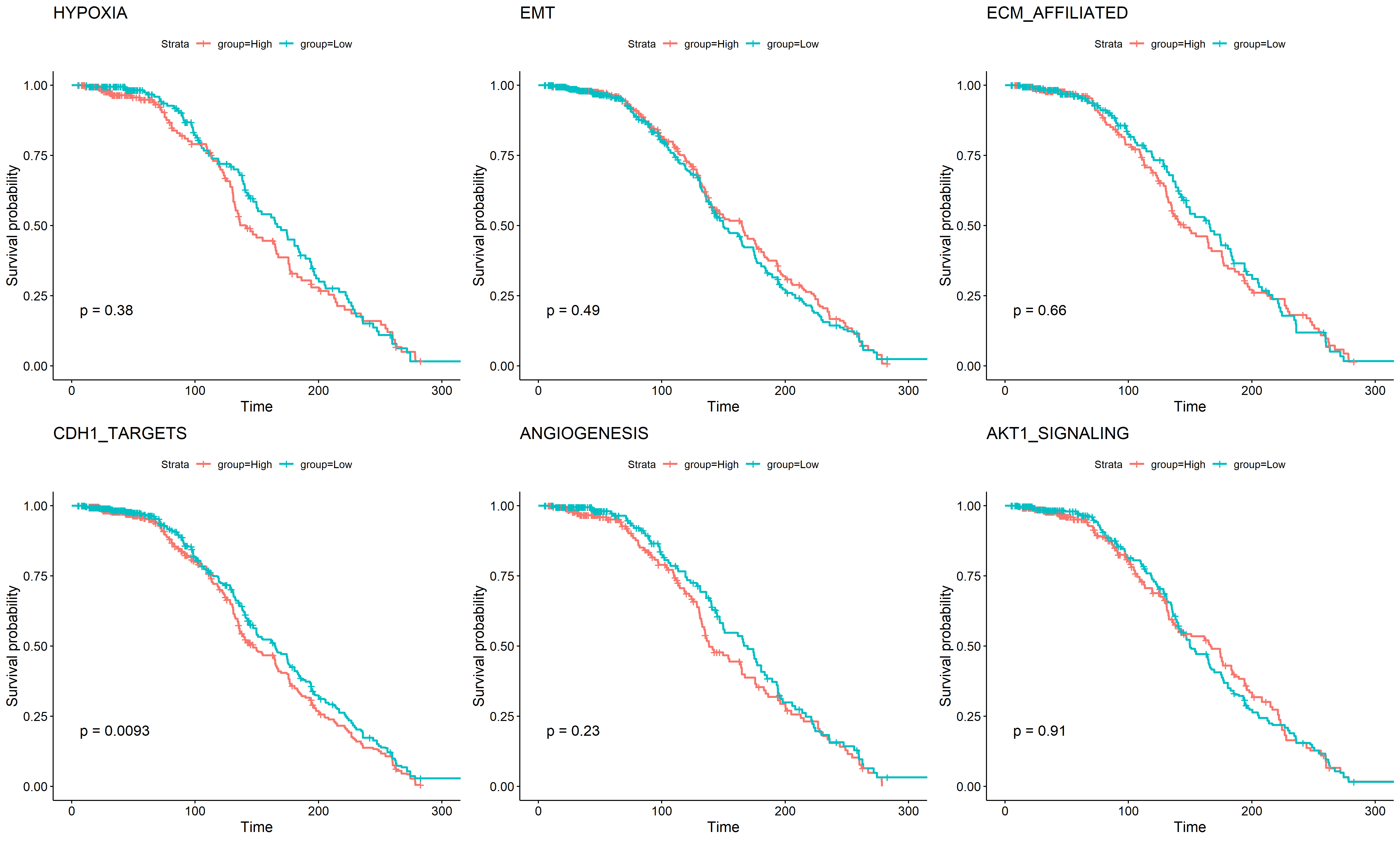
library(survival)library(survminer)gs <- split(dat3,dat3$geneset)for (i in 1:length(gs)) {x=gs[[i]]x <- filter(x,abs(value) >= 0.1)x$group <- ifelse(x$value >= 0.1, "High","Low")table(x$group)mySurv_OS=with(x,Surv(OS_MONTHS, OS_STATUS))sfit=survfit(mySurv_OS~group,data=x)print(ggsurvplot(sfit,pval =TRUE,title=x$geneset))ggsave(paste0(i,'.png'))}
A. 根据是否化疗挑选病人
B. 根据 claudin subtype 挑选病人
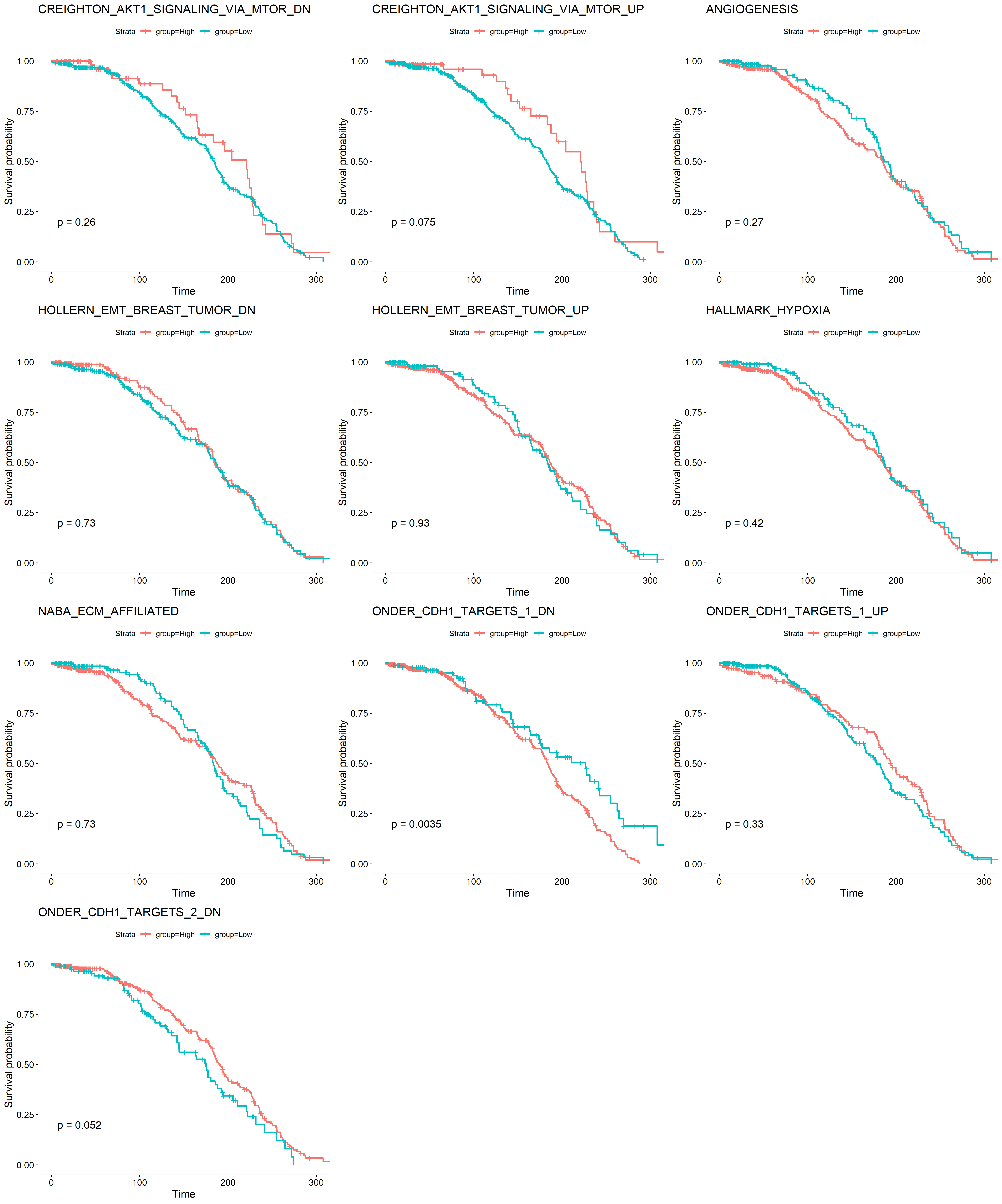
合并具有UP和DOWN的基因集再画图
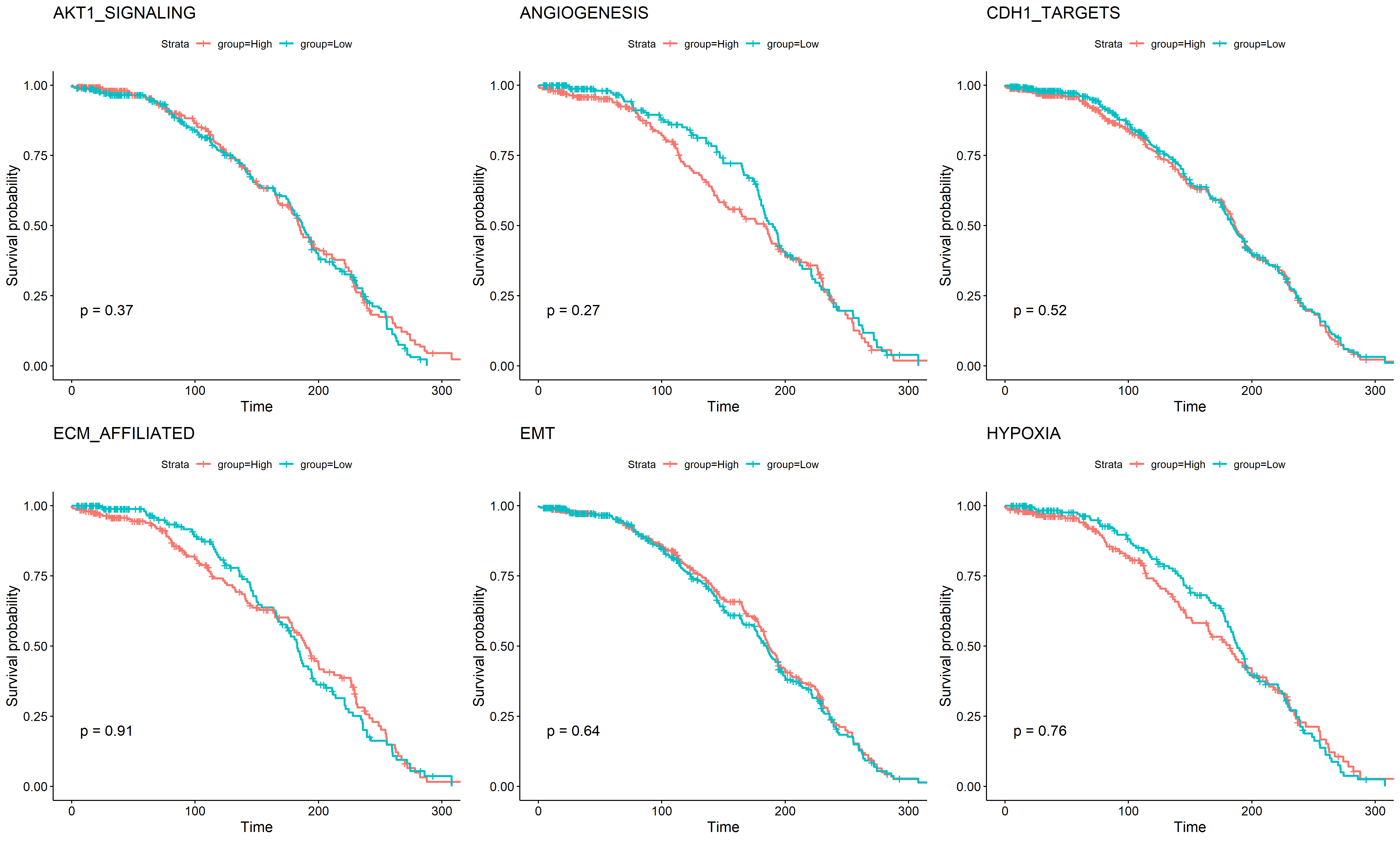
dat3$geneset <- ifelse(grepl("AKT1",dat3$geneset)==TRUE,"AKT1_SIGNALING",ifelse(grepl("EMT",dat3$geneset)==TRUE,"EMT",ifelse(grepl("ECM",dat3$geneset)==TRUE,"ECM_AFFILIATED",ifelse(grepl("HYPOXIA",dat3$geneset)==TRUE,"HYPOXIA",ifelse(grepl("ANGIOGENESIS",dat3$geneset)==TRUE,"ANGIOGENESIS","CDH1_TARGETS")))))
### 根据是否化疗挑选病人 ###table(dat3$geneset)## AKT1_SIGNALING ANGIOGENESIS CDH1_TARGETS ECM_AFFILIATED EMT HYPOXIA# 792 396 2376 396 792 396### 根据 claudin subtype 挑选病人###table(dat3$geneset)## AKT1_SIGNALING ANGIOGENESIS CDH1_TARGETS ECM_AFFILIATED# 796 398 2388 398# EMT HYPOXIA# 796 398
gs2 <- split(dat3,dat3$geneset)for (i in 1:length(gs2)) {x=gs2[[i]]x$group <- ifelse(x$value > median(x$value), "High","Low")table(x$group)mySurv_OS=with(x,Surv(OS_MONTHS, OS_STATUS))sfit=survfit(mySurv_OS~group,data=x)print(ggsurvplot(sfit,pval =TRUE,title=x$geneset))ggsave(paste0(i,'_1.png'))}
A. 根据是否化疗挑选病人
B. 根据 claudin subtype 挑选病人

若有收获,就点个赞吧
0 人点赞
