数据准备
1. 表达矩阵
- 将行名转换为 gene symbol
- 数值log2后取小数点后两位
- 锁定11个基因
suppressPackageStartupMessages(library(org.Hs.eg.db))suppressPackageStartupMessages(library(AnnotationDbi))expr <- read.table('./TCGA-HNSC.htseq_counts.tsv',header = T,sep='\t')dim(expr)# [1] 60488 547expr[1:4,1:4]# Ensembl_ID TCGA.BB.4224.01A TCGA.H7.7774.01A TCGA.CV.6943.01A# 1 ENSG00000000003.13 11.127994 11.420487 11.39178# 2 ENSG00000000005.5 1.584963 0.000000 0.00000# 3 ENSG00000000419.11 10.650154 10.724514 10.68825# 4 ENSG00000000457.12 10.055282 9.651052 9.84235enb <- as.character(expr[,1])enb_new <- c()for (i in 1:length(enb)) {enb_new[i] <- strsplit(enb[i],"\\.")[[1]][1]}enb_new <- unique(enb_new)symb <- AnnotationDbi::select(org.Hs.eg.db,enb_new,"SYMBOL","ENSEMBL")todelet <- match(symb[,1],enb_new )expr[,1] <- enb_newexpr <- expr[todelet,]expr[,1] <- symb[,2]gs <- expr$Hugo_Symbolexpr <- expr[,-1]rownames(expr) <- gsexpr <- log2(expr+1)expr <- apply(expr, 2, function(x){as.numeric(format(x,digits = 2))})save(expr,file='expression.Rdata')
2. 临床数据
- 将行名形式与表达矩阵统一
clin <- read.table('./TCGA-HNSC.GDC_phenotype.tsv',header = T,sep='\t')summary(clin)clin[1:4,1:4]clin$submitter_id.samples = gsub('-','.',clin$submitter_id.samples)row.names(clin) <- clin[,1]save(clin,file='clinical.Rdata')
3. 生存数据
surv <- read.table('./TCGA-HNSC.survival.tsv',header = T,sep='\t')
4. 整合数据
- 锁定11个基因
- 整合
clin和surv
geneselected <- expr[c("CHAC2","CLEC9A","GNG10","JCHAIN","KLRB1","NOG","OLR1","PRELID2","SYT1","VWCE","ZNF443"),]phe <- clin[,c("submitter_id.samples","age_at_initial_pathologic_diagnosis","clinical_stage")]colnames(phe) <- c("ID","age","stage")row.names(phe) <- phe[,1]row.names(surv) <- surv[,1]phe <- cbind(phe,surv[,c("OS.time","OS")])phe <- phe[phe$ID %in% surv$sample,]surv <- surv[surv$sample %in% phe$ID,]phe <- cbind(phe,surv[,c("OS.time","OS")])## 去除 stage 中的空白值phe <- phe[phe$stage!="",]geneselected <- geneselected[,colnames(geneselected) %in% phe$ID]## 更改数据框中所有列的属性,和示例数据一致phe$age <- as.numeric(phe$age)phe$stage <- as.character(phe$stage)phe$OS.time <- as.numeric(phe$OS.time)phe$OS <- as.numeric(phe$OS)
coxph
mySurv=with(phe,Surv(OS.time, OS))cox_results <-apply(geneselected, 1 ,function(gene){group=ifelse(gene>median(gene),'high','low')survival_dat <- data.frame(group=group,stage=phe$stage,age=phe$age,stringsAsFactors = F)m=coxph(mySurv ~ age + stage+ group, data = survival_dat)beta <- coef(m)se <- sqrt(diag(vcov(m)))HR <- exp(beta)HRse <- HR * setmp <- round(cbind(coef = beta, se = se, z = beta/se, p = 1 - pchisq((beta/se)^2, 1),HR = HR, HRse = HRse,HRz = (HR - 1) / HRse, HRp = 1 - pchisq(((HR - 1)/HRse)^2, 1),HRCILL = exp(beta - qnorm(.975, 0, 1) * se),HRCIUL = exp(beta + qnorm(.975, 0, 1) * se)), 3)return(tmp['grouplow',])})cox_results=t(cox_results)table(cox_results[,4]<0.05)## FALSE TRUE# 10 1cox_results[cox_results[,4]<0.05,]# coef se z p HR HRse HRz HRp HRCILL HRCIUL# 0.398 0.193 2.058 0.040 1.489 0.288 1.698 0.090 1.019 2.176
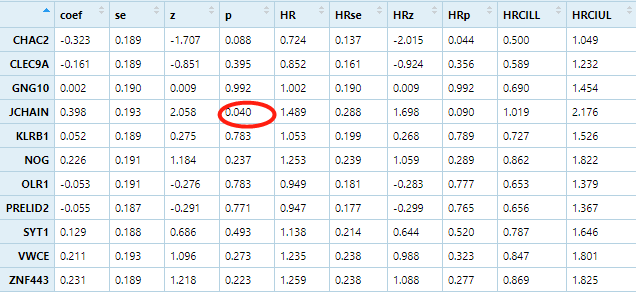
结果只有一个基因是显著的。
同时还有另一个问题(纠缠了大半天
当矩阵是全部基因的 expr 时,一直会出现报错,目前还未解决。
也是因为这个报错,删去了satge有空白值的项、更改了数据框每一列的属性,不过依旧报错,没有结果文件产生。
换成目标的11个基因的表达矩阵后,没有出现报错。

若有收获,就点个赞吧
0 人点赞
