本文来自“单细胞组学”公众号 原文链接:https://mp.weixin.qq.com/s/vZFO2caZXNT0DWqLYvJe4g
背景:
单细胞的基因表达存在方差异性,基因表达在细胞间的方差和平均表达量呈正相关。需要选择合适的统计方法,减小高变基因选择的偏差。
统计方法比较:
大致分为三大类:
- 根据基因平均表达量和方差的关系,建立回归模型,使得表达差异独立于平均表达量;基于该原理的方法有scLVM、scran 和Seurat的vst [1,2,3]。
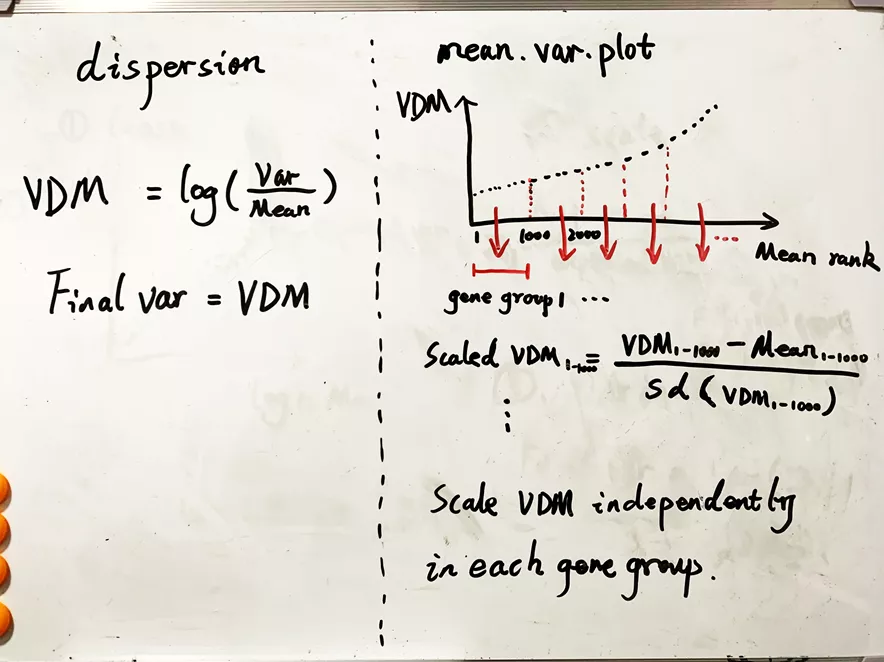
- 用方差或者标准差除以平均数的值作为细胞间差异系数(VDM),再用模型量化,抵消平均数的影响。如Seurat的mean.var.plot. 先根据表达平均数划分若干个区,然后每个区的基因独立做z-score量化。
- 其他方法。如BASICS的贝叶斯分层模型(略)[4]。
结论:
- 高变基因集中在低表达基因。
2. 高变基因有较高的零值比例。
3. 大多数高变基因的方差大于负二项模型预估的方差。
单细胞数据的重要特征:均值与方差的关系可以用泊松模型预估。
单细胞原始数据标准化后,需要选择一定数量的高变基因(high variable genes,1000~3000),减轻后续降维计算难度,同时保留生物差异特征。因为单细胞表达矩阵的稀疏性和基因表达的方差异性,高表达的基因往往有更高的方差,直接用方差计算细胞间差异,会造成偏差。去年一月份Valentine Svensson写给NBT的一篇文章用负二项模型(gamma泊松模型)预估平均值和方差的关系,然后用多个数据集证明droplet数据过多的零值不是技术因素,而是生物本身的差异[5]。下图展示单一细胞数据集(HEK293T)和多种细胞数据集(PBMC)的基因分布差异。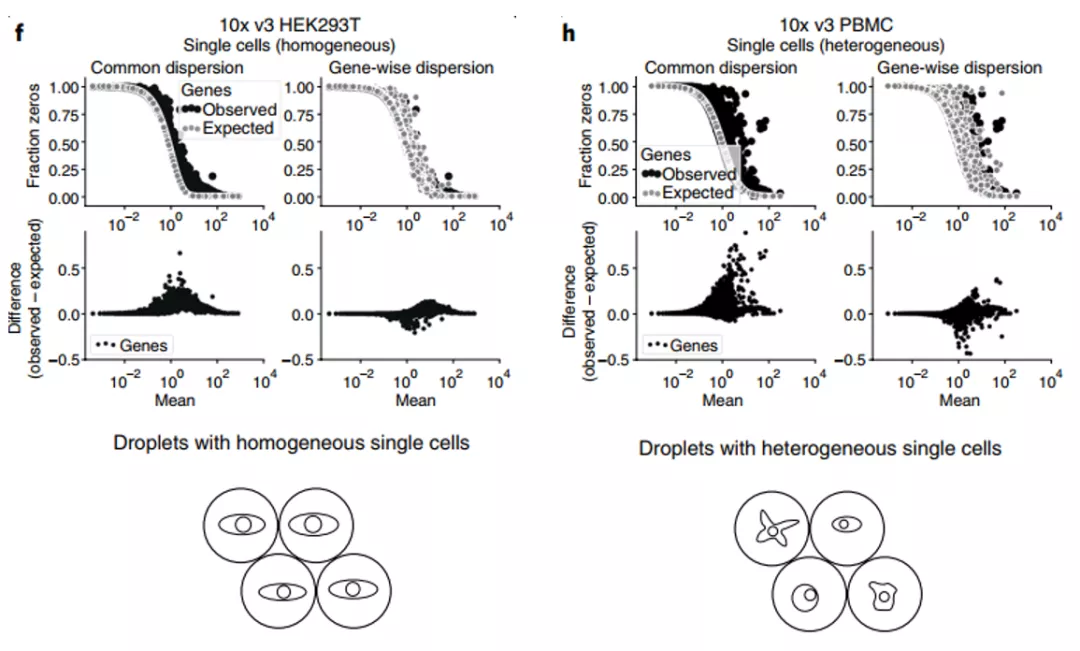
我们可以用多种函数来做拟合。比如python的scipy.optimize.curve_fit模块的fit_curve方法,和R的广义线性模型的函数glm。glm也是我们上期解析sctransform时用到的函数,用来预估一个基因在不同细胞的表达量和每个细胞的总UMI数(测序深度)之间的关系。下面的代码是用python的fit_curve得到均值和方差的系数phi:
library(reticulate)testdata <- pbmc@assays$RNA@countsMean <- Matrix::rowMeans(testdata)Var <- apply(testdata, 1, var)x <- as.vector(Mean)y <- as.vector(Var)#plot(log10(x), log10(y))##fit formula by python scipy.optimize.curve_fitphi<- fit_curve(x, y)expected_var <- x + phi*x^2#red line is expected variance.
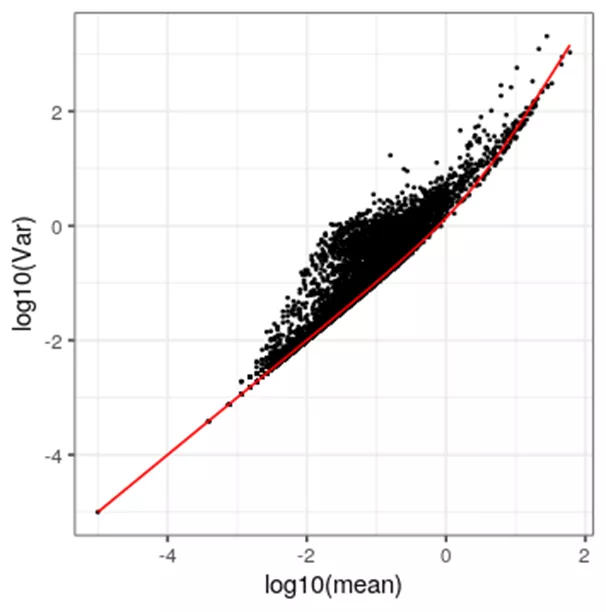
可以看出均值和方差呈log的正相关,均值中间有多个基因高于预期的方差。后面我们可以看到这些高出来的点大多是我们要寻找的高变基因。
Seurat三种高变基因选择方法的比较
Seurat的FindVariableFeatures函数中有三种方法可供选择:
- vst;
- mean.var.plot;
- dispersion.
简而言之,vst用局部加权回归(loess)预估均值和方差的关系得到期望的方差值,然后z-score量化表达矩阵,最后计算方差;dispersion比较简单粗暴,用方差/均值(VDM);mean.var.plot先计算每个基因的VDM,然后根据平均表达量对每个基因由低到高进行分区,在每个分区内独立计算VDM的z-score。如小白板所示。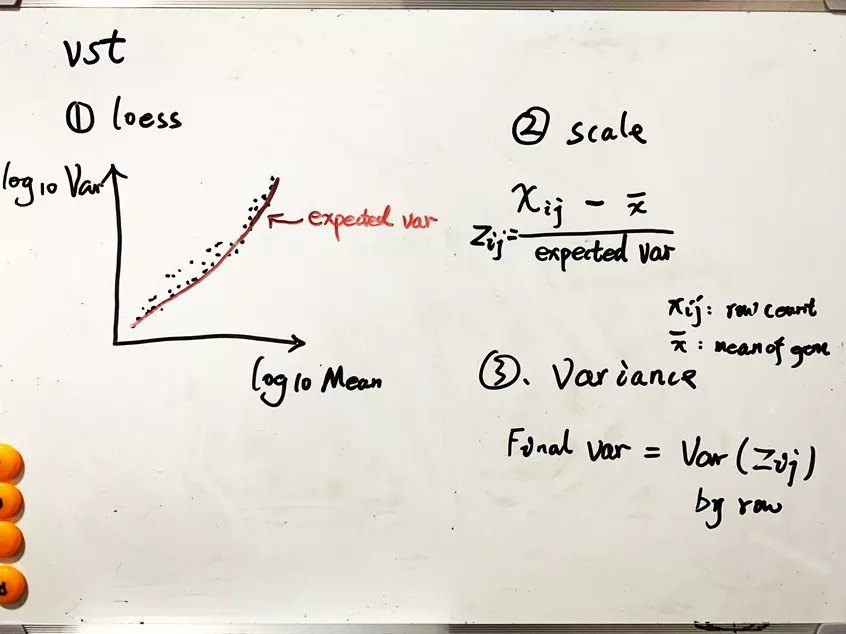
高变基因的特征
1. 集中在低表达基因。
Seurat三种方法都是如此。最低表达的基因分区1已经包括了1500/2000以上的高变基因。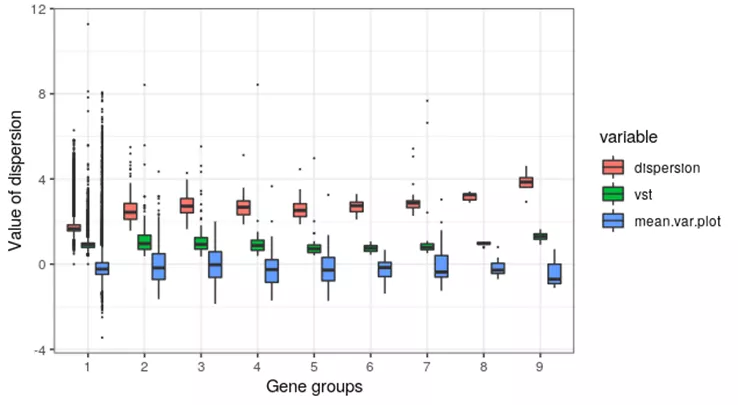
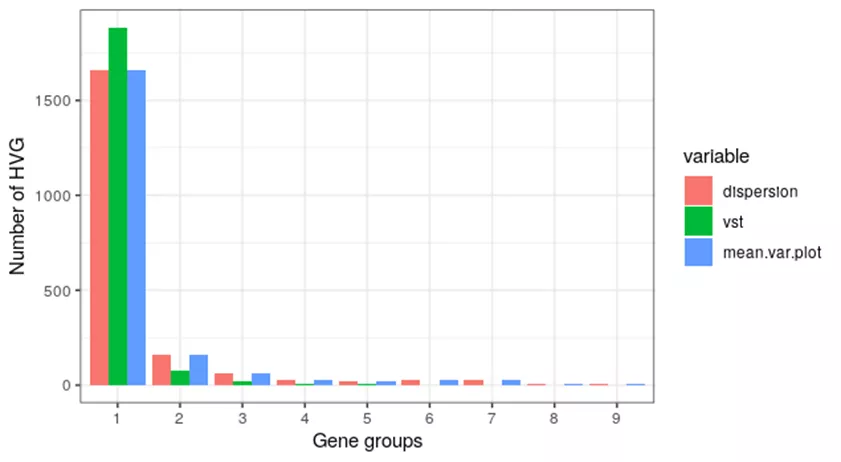
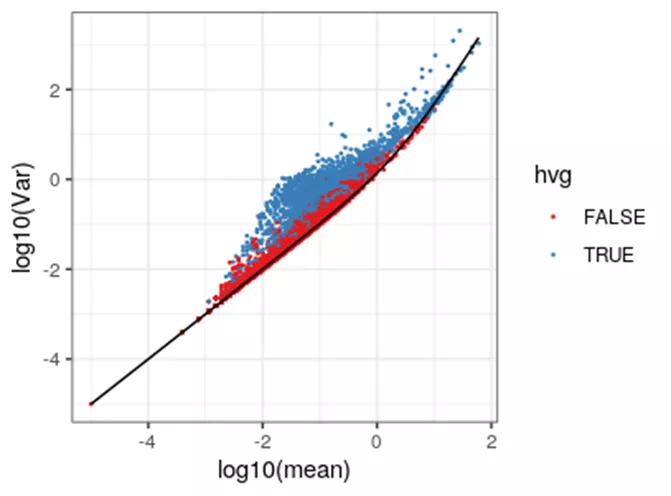
- 大多数高变基因的方差大于负二项模型预估的方差。
3. 高变基因有较高的零值比例。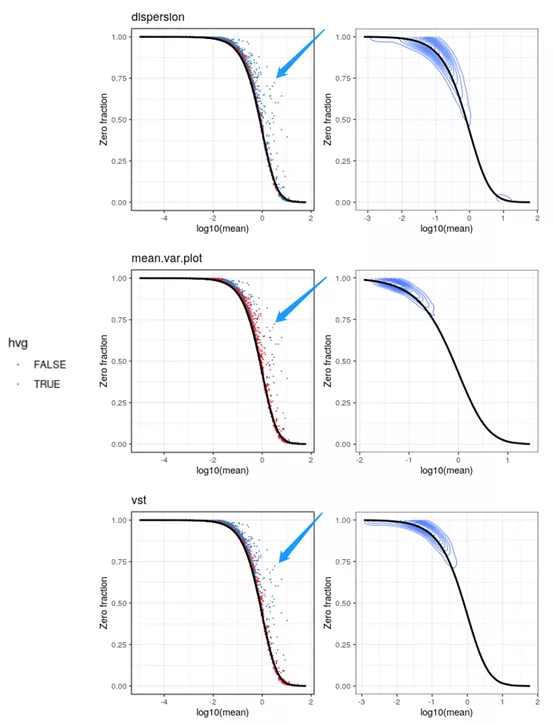
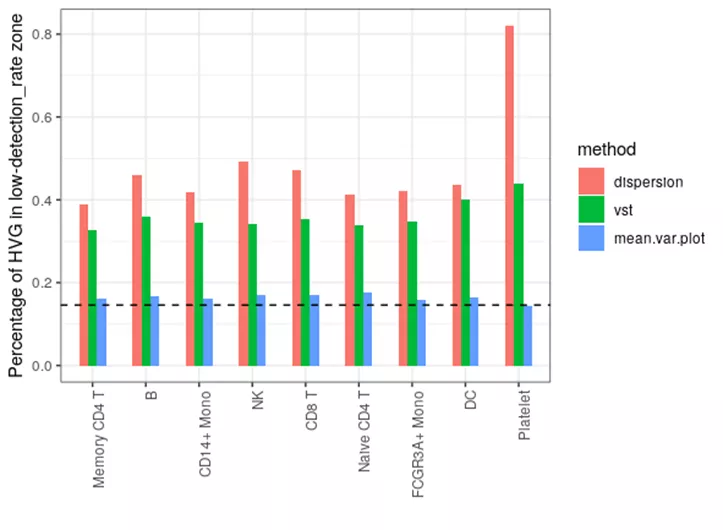
三种方法的高变基因在高零值比例的基因区域分布存在差异。dispersion的零值比例最高,其次是vst,而mean.var.plot的零值比例只稍微高出整体(2000/所有基因数)。高变基因具有较高的零值比例,说明很多高变基因只在小部分细胞群表达。
Seurat三种方法的R基础函数实现以及负二项分布模型代码可以在我的github上查看[6,7]。
下期预告:空间转录组的高变基因鉴定方法 (SpatialDE, Trendsceek, Splotch)
参考链接:
[1]. Buettner F, Natarajan K N, Casale F P, etal. Computational analysis of cell-to-cell heterogeneity in single-cellRNA-sequencing data reveals hidden subpopulations of cells[J]. Naturebiotechnology, 2015, 33(2): 155-160.
[2]. Lun, Aaron TL, Karsten Bach, and John C.Marioni.”Pooling across cells to normalize single-cell RNA sequencingdata withmany zero counts.” Genome biology 17.1 (2016): 75.
[3]. Butler A, Hoffman P, Smibert P, et al.Integrating single-cell transcriptomic data across different conditions,technologies, and species[J]. Nature biotechnology, 2018, 36(5): 411-420.
[4]. Vallejos, Catalina A., John C. Marioni,andSylvia Richardson. “BASiCS: Bayesian analysis of single-cellsequencingdata.” PLoS Comput Biol 11.6 (2015): e1004333.
[5]. Svensson V. Droplet scRNA-seq is notzero-inflated[J]. Nature Biotechnology, 2020, 38(2): 147-150.
[6]. https://github.com/Zhihao-Huang/SingleCellOmics/blob/master/base_FindVar.R
[7]. https://github.com/Zhihao-Huang/SingleCellOmics/blob/master/density_nb_fit.R

若有收获,就点个赞吧
0 人点赞
